

Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists
- PMID: 28286377
- PMCID: PMC5327760
- DOI: 10.1155/2017/7018393
Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists
Abstract
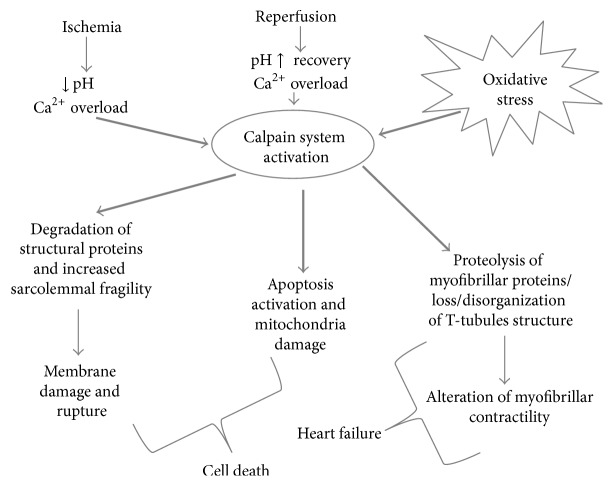
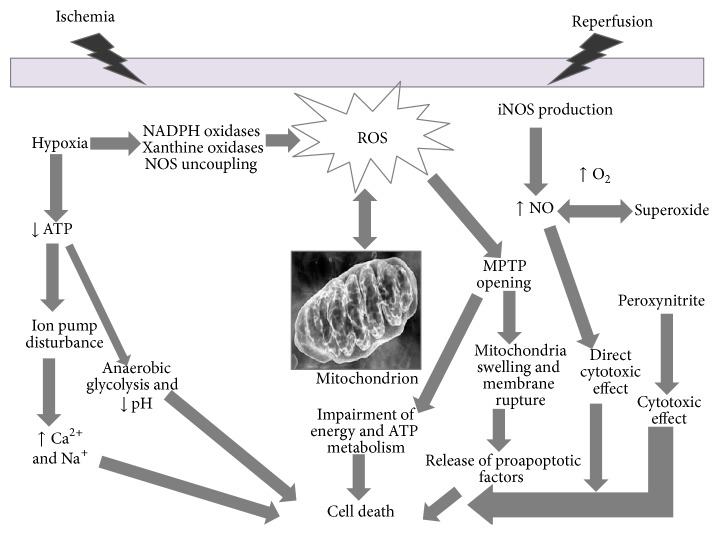
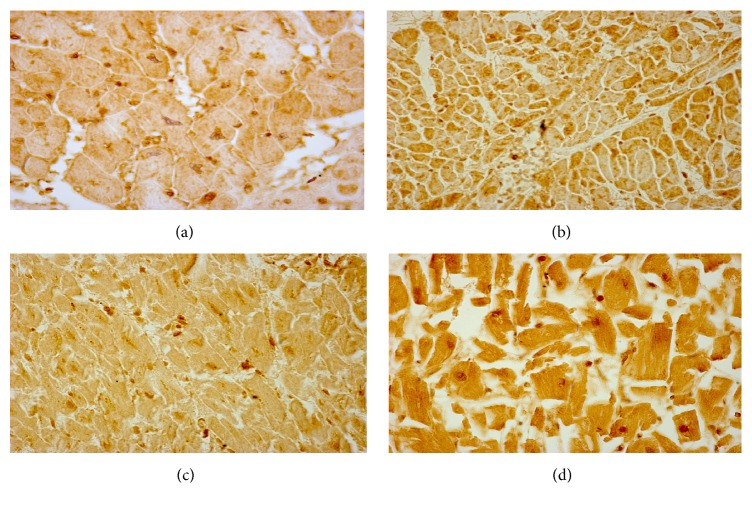
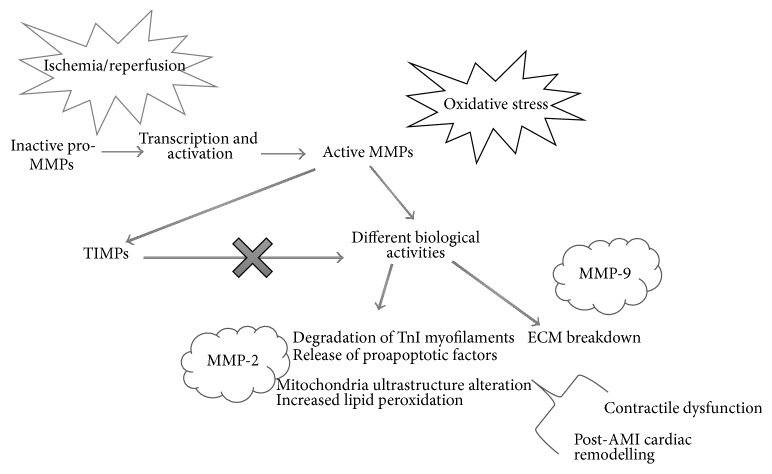
Acute myocardial infarction (AMI) is a leading cause of morbidity and mortality. Reperfusion strategies are the current standard therapy for AMI. However, they may result in paradoxical cardiomyocyte dysfunction, known as ischemic reperfusion injury (IRI). Different forms of IRI are recognized, of which only the first two are reversible: reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction, and lethal myocardial reperfusion injury. Sudden death is the most common pattern for ischemia-induced lethal ventricular arrhythmias during AMI. The exact mechanisms of IRI are not fully known. Molecular, cellular, and tissue alterations such as cell death, inflammation, neurohumoral activation, and oxidative stress are considered to be of paramount importance in IRI. However, comprehension of the exact pathophysiological mechanisms remains a challenge for clinicians. Furthermore, myocardial IRI is a critical issue also for forensic pathologists since sudden death may occur despite timely reperfusion following AMI, that is one of the most frequently litigated areas of cardiology practice. In this paper we explore the literature regarding the pathophysiology of myocardial IRI, focusing on the possible role of the calpain system, oxidative-nitrosative stress, and matrix metalloproteinases and aiming to foster knowledge of IRI pathophysiology also in terms of medicolegal understanding of sudden deaths following AMI.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Mozaffarian D., Benjamin E. J., Go A. S., et al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–e322. - PubMed
-
- American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, O'Gara P. T., et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology. 2013;61:e78–e140. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
