

Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer
- PMID: 28287083
- PMCID: PMC5355811
- DOI: 10.1038/ncomms14768
Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer
Abstract
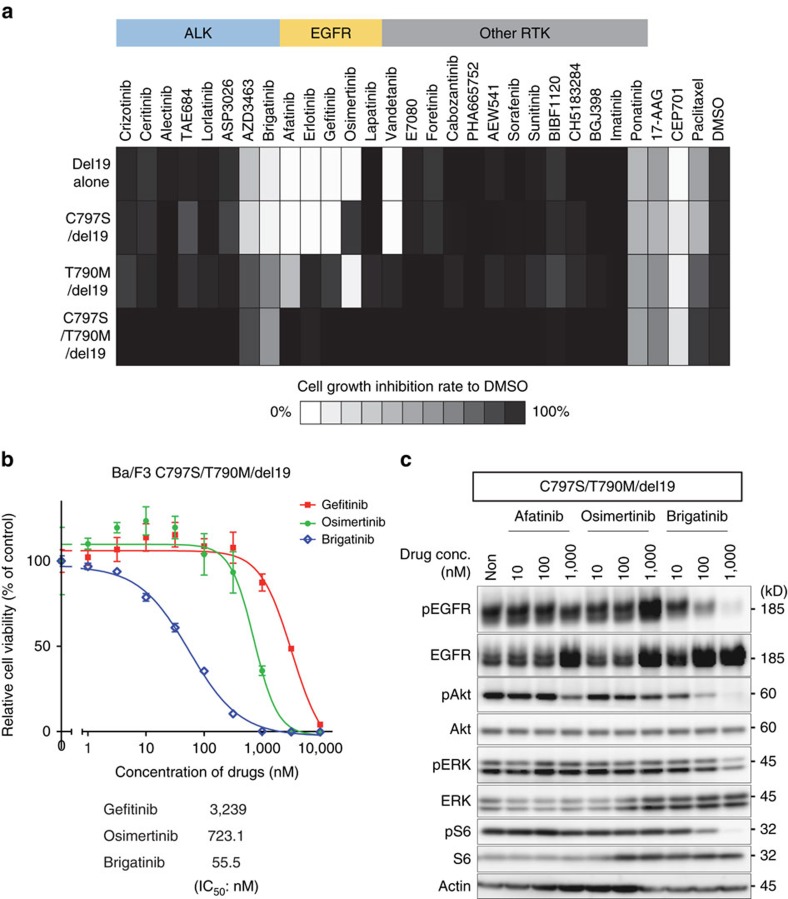
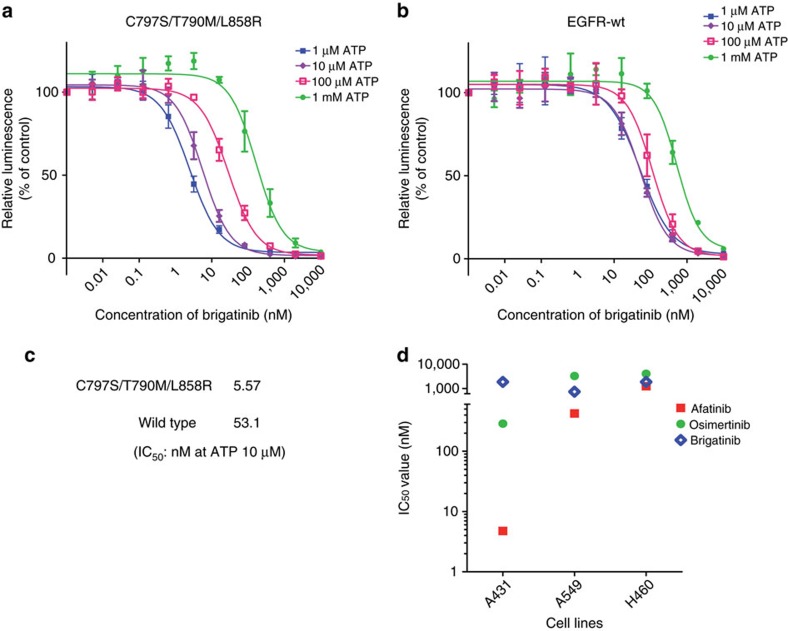
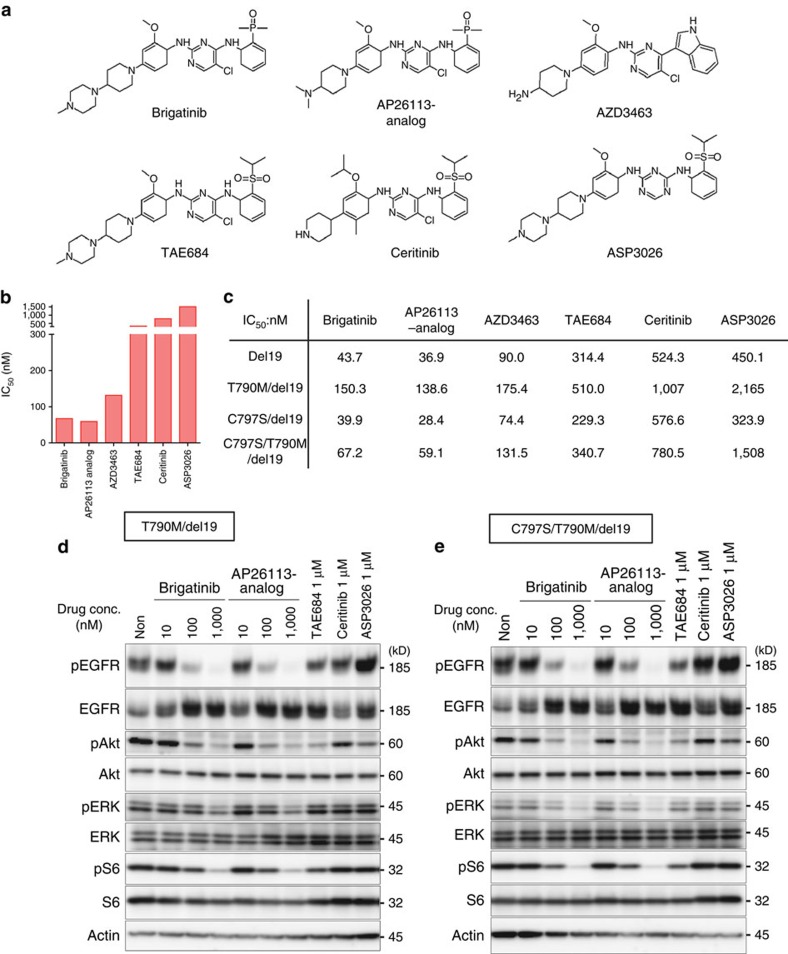
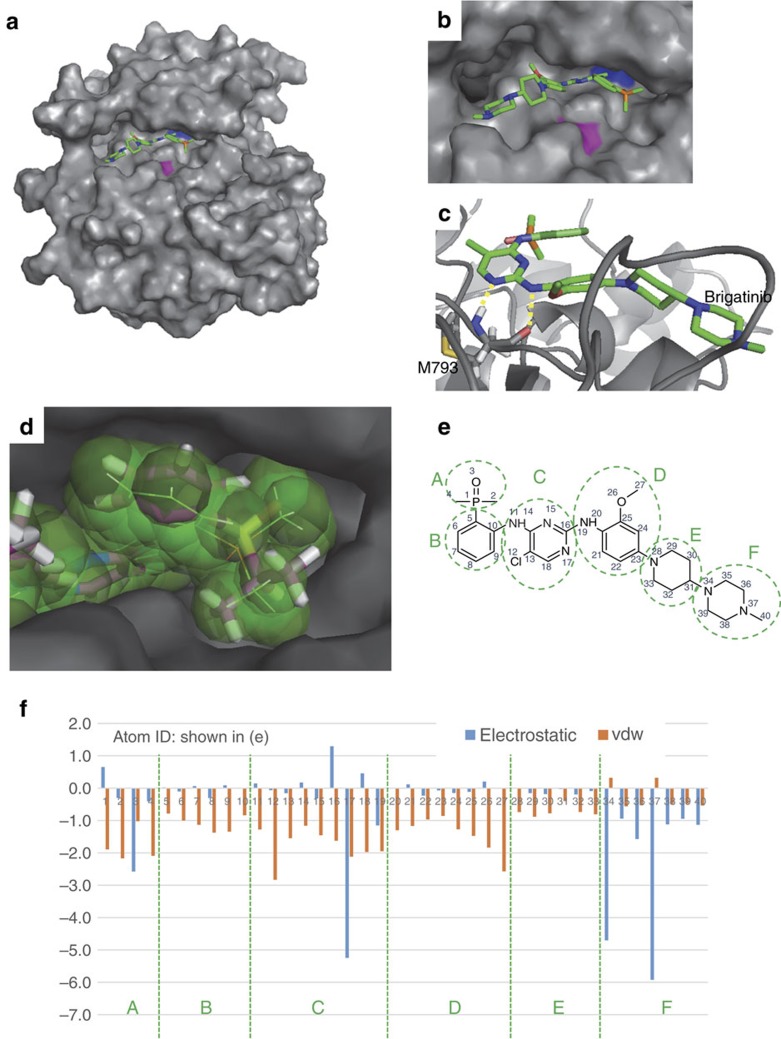
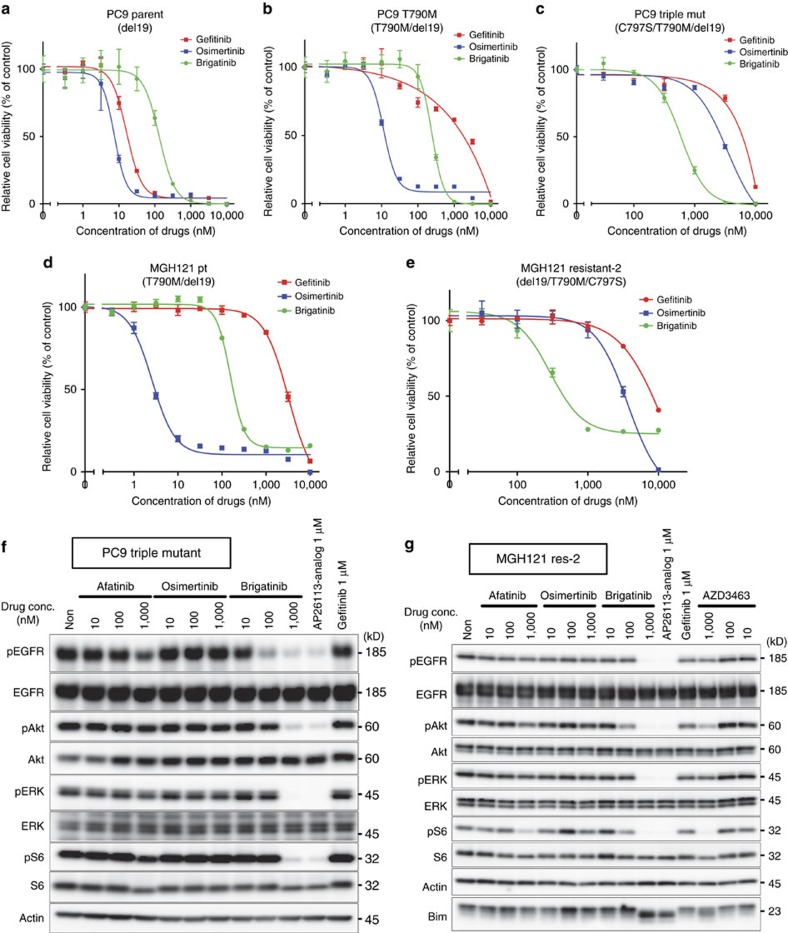
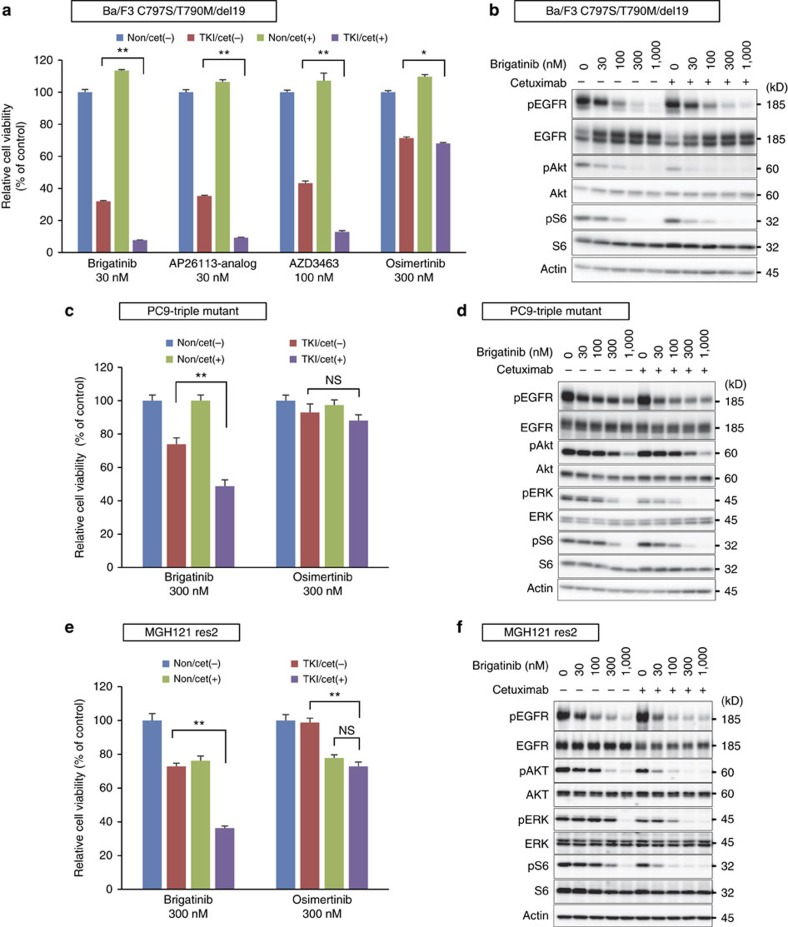
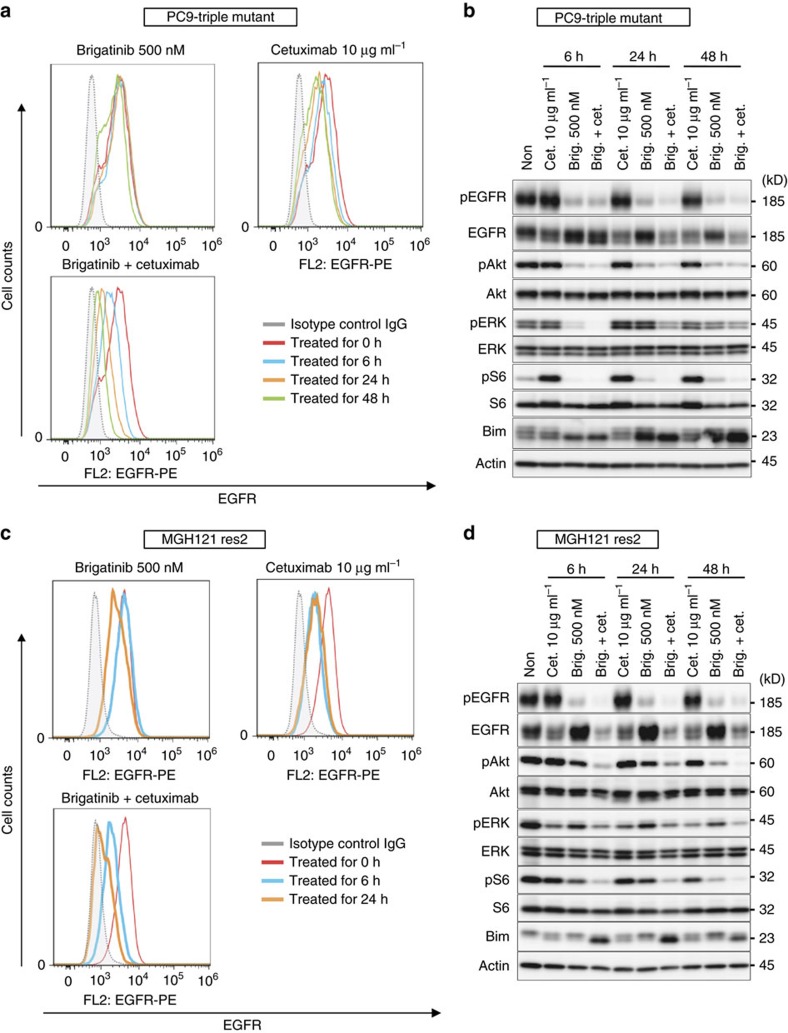
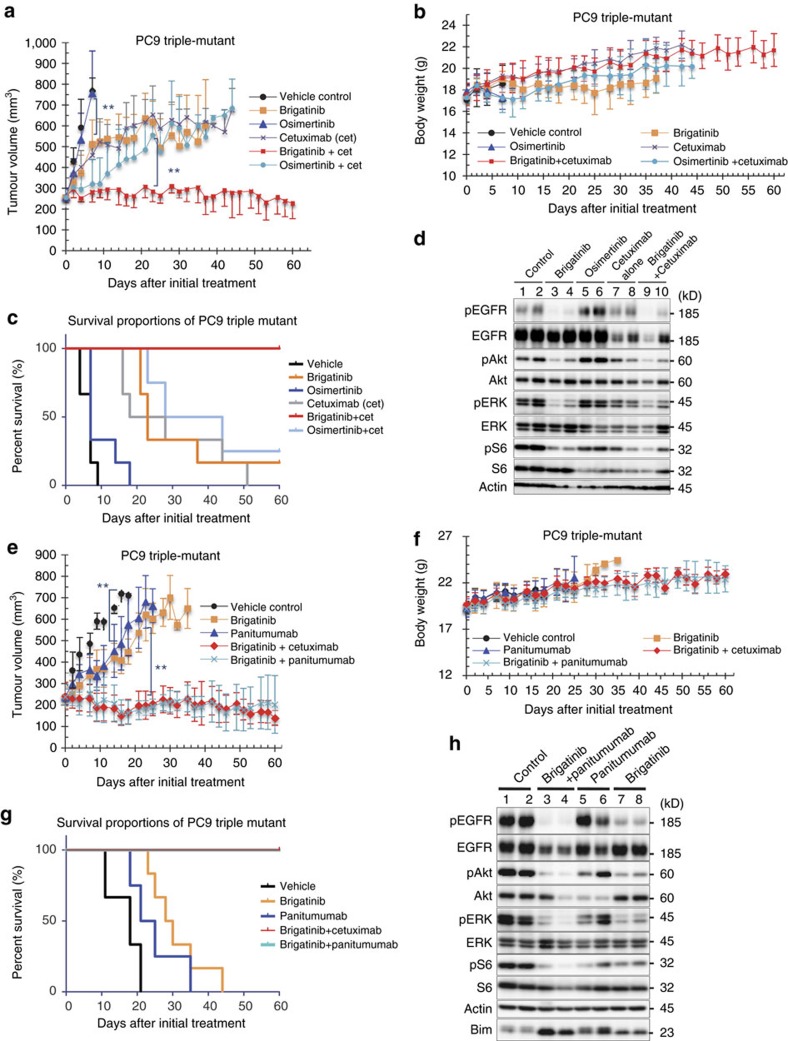
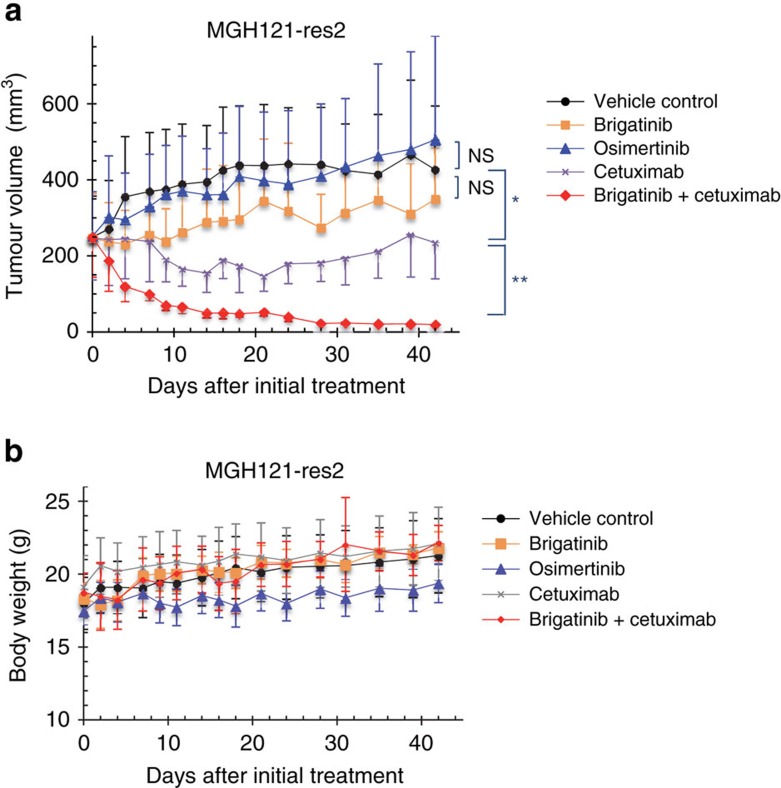
Osimertinib has been demonstrated to overcome the epidermal growth factor receptor (EGFR)-T790M, the most relevant acquired resistance to first-generation EGFR-tyrosine kinase inhibitors (EGFR-TKIs). However, the C797S mutation, which impairs the covalent binding between the cysteine residue at position 797 of EGFR and osimertinib, induces resistance to osimertinib. Currently, there are no effective therapeutic strategies to overcome the C797S/T790M/activating-mutation (triple-mutation)-mediated EGFR-TKI resistance. In the present study, we identify brigatinib to be effective against triple-mutation-harbouring cells in vitro and in vivo. Our original computational simulation demonstrates that brigatinib fits into the ATP-binding pocket of triple-mutant EGFR. The structure-activity relationship analysis reveals the key component in brigatinib to inhibit the triple-mutant EGFR. The efficacy of brigatinib is enhanced markedly by combination with anti-EGFR antibody because of the decrease of surface and total EGFR expression. Thus, the combination therapy of brigatinib with anti-EGFR antibody is a powerful candidate to overcome triple-mutant EGFR.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Giaccone G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. J. Clin. Oncol. 23, 3235–3242 (2005). - PubMed
-
- Maemondo M. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388 (2010). - PubMed
-
- Mok T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009). - PubMed
-
- Mitsudomi T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128 (2010). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
