

Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1α-dependent glycolysis
- PMID: 28287112
- PMCID: PMC6079089
- DOI: 10.1038/cmi.2017.5
Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1α-dependent glycolysis
Abstract
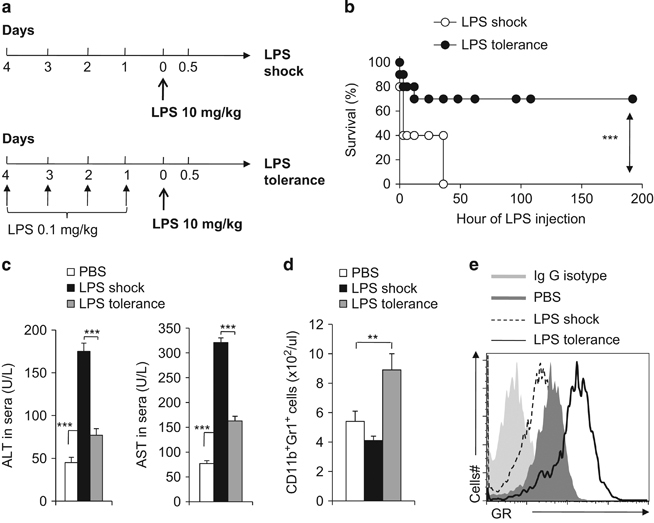
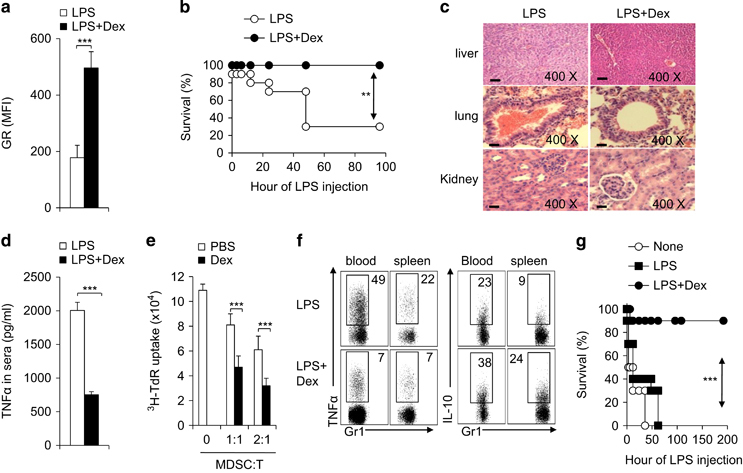
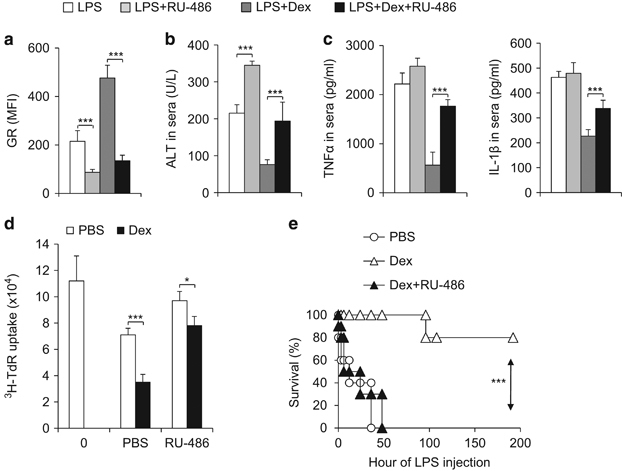
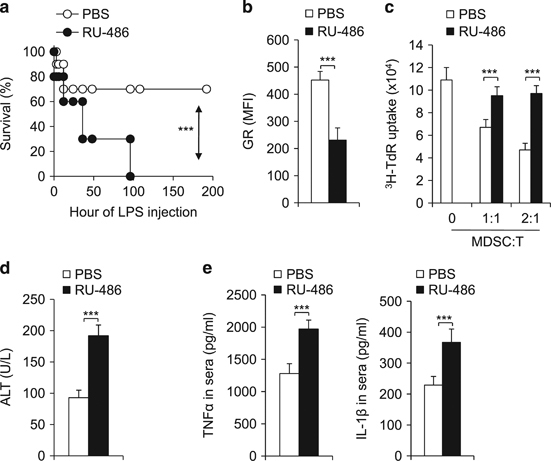
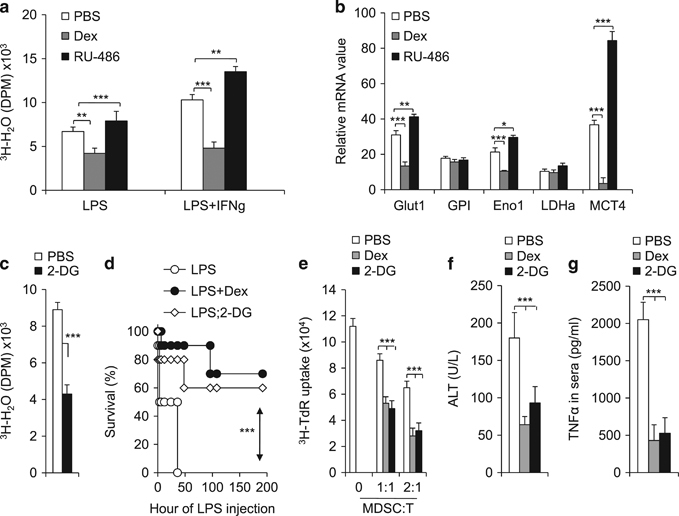
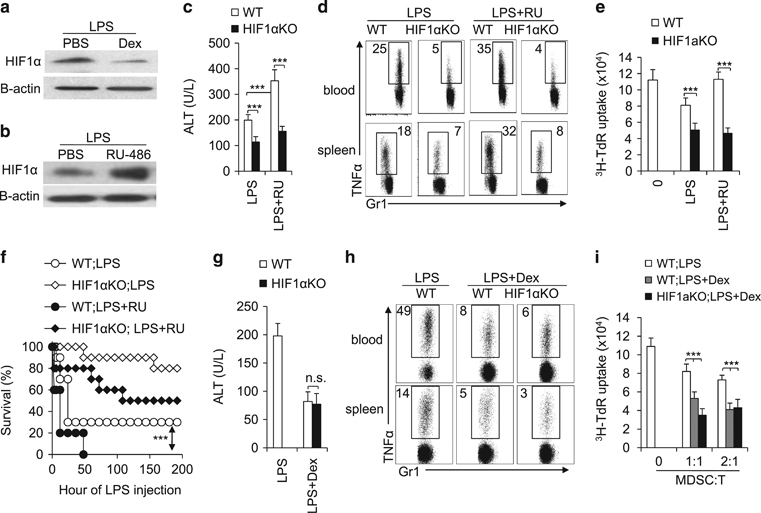
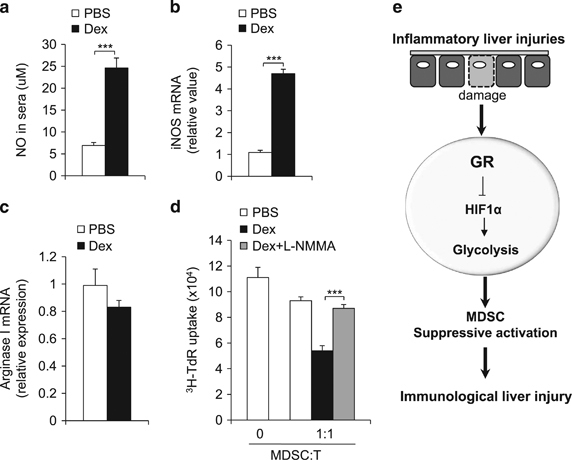
Immunomodulatory signaling imposes tight regulations on metabolic programs within immune cells and consequentially determines immune response outcomes. Although the glucocorticoid receptor (GR) has been recently implicated in regulating the function of myeloid-derived suppressor cells (MDSCs), whether the dysregulation of GR in MDSCs is involved in immune-mediated hepatic diseases and how GR regulates the function of MDSCs in such a context remains unknown. Here, we revealed the dysregulation of GR expression in MDSCs during innate immunological hepatic injury (IMH) and found that GR regulates the function of MDSCs through modulating HIF1α-dependent glycolysis. Pharmacological modulation of GR by its agonist (dexamethasone, Dex) protects IMH mice against inflammatory injury. Mechanistically, GR signaling suppresses HIF1α and HIF1α-dependent glycolysis in MDSCs and thus promotes the immune suppressive activity of MDSCs. Our studies reveal a role of GR-HIF1α in regulating the metabolism and function of MDSCs and further implicate MDSC GR signaling as a potential therapeutic target in hepatic diseases that are driven by innate immune cell-mediated systemic inflammation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
