

Myocardial Ischemic Postconditioning Promotes Autophagy against Ischemia Reperfusion Injury via the Activation of the nNOS/AMPK/mTOR Pathway
- PMID: 28287478
- PMCID: PMC5372630
- DOI: 10.3390/ijms18030614
Myocardial Ischemic Postconditioning Promotes Autophagy against Ischemia Reperfusion Injury via the Activation of the nNOS/AMPK/mTOR Pathway
Abstract
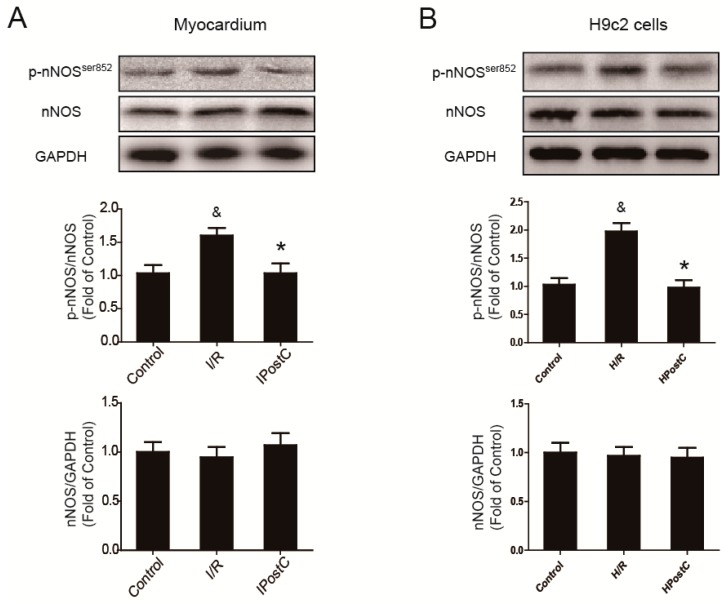
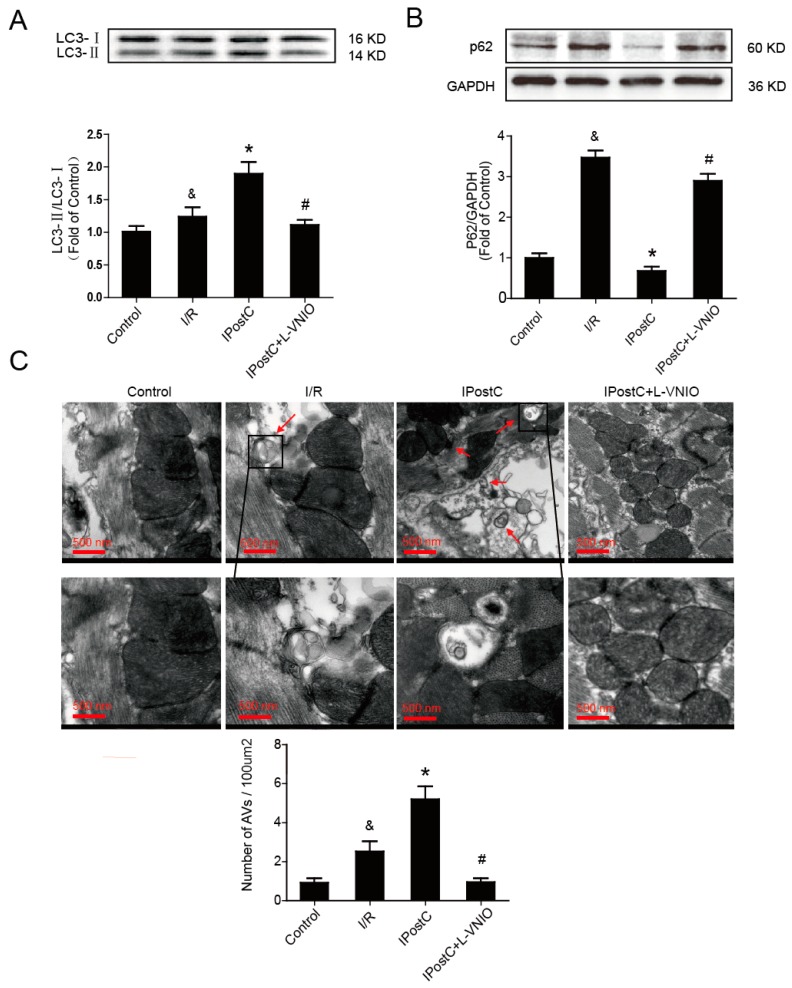
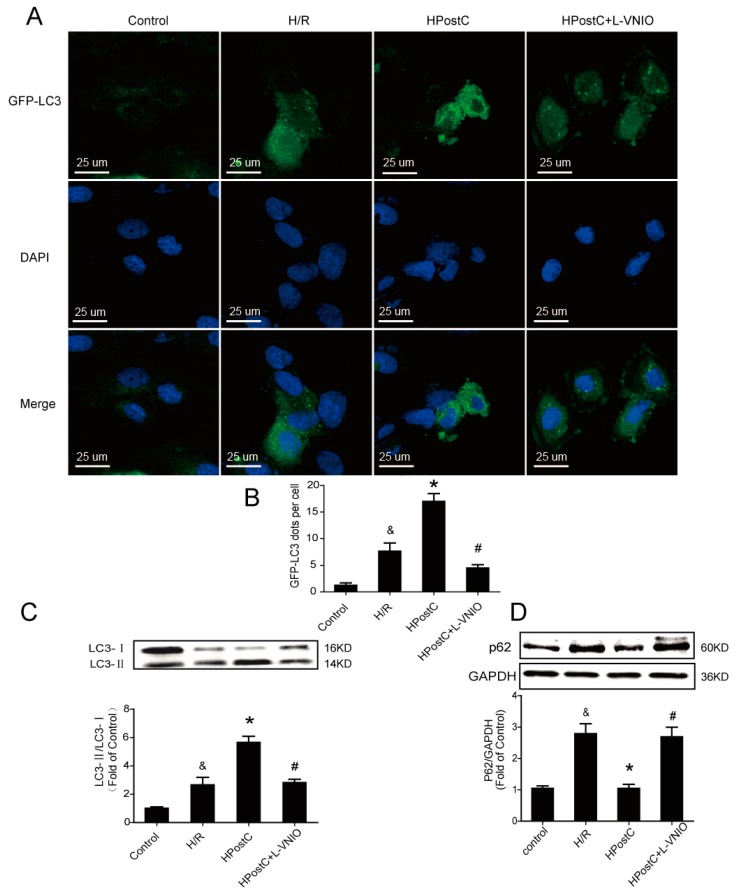
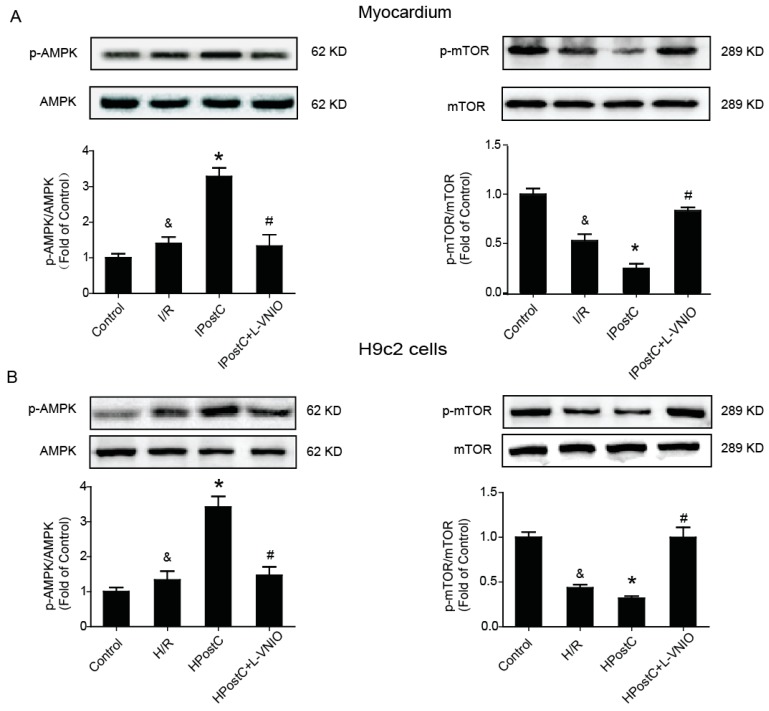
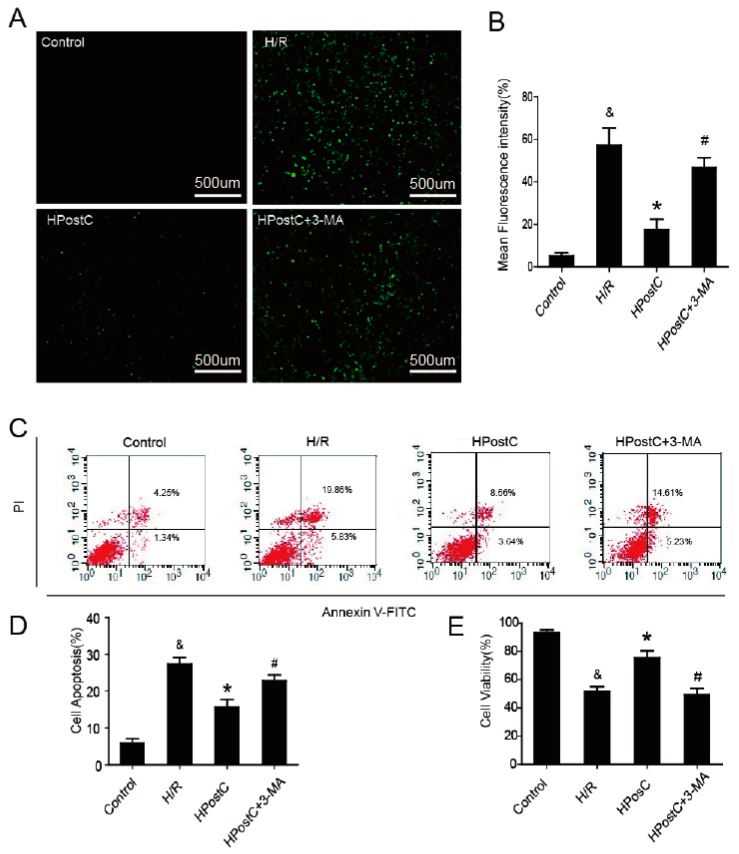
Autophagy participates in the progression of many diseases, comprising ischemia/ reperfusion (I/R). It is reported that it is involved in the protective mechanism of ischemic postconditioning (IPostC). According to research, neuronal nitric oxide synthase (nNOS) is also involved in the condition of I/R and IPostC. However, the relationship between nNOS, autophagy and IPostC has not been previously investigated. We hypothesize that IPostC promotes autophagy activity against I/R injury partially through nNOS-mediated pathways. Mouse hearts were subjected to I/R injury through the ligation of the left anterior descending coronary artery. H9c2 cells were subjected to hypoxia/reoxygenation (H/R) in vitro. IPostC, compared with I/R, restored nNOS activity, increased the formation of autophagosome and restored the impaired autophagic flux, thus autophagic activity was raised markedly. IPostC increased adenosine monophosphate-activated protein kinase (AMPK) phosphorylation and suppressed mammalian target of rapamycin (mTOR), but a selective nNOS inhibitor abolished those effects. Similar effects of IPostC were demonstrated in H9c2 cells in vitro. IPostC decreased infarct size and preserved most of the normal structure. The level of reactive oxygen species (ROS) and cell apoptosis were reduced by IPostC with improved cell viability and mitochondrial membrane potential. However, an autophagy inhibitor suppressed the protective effects. These results suggest that IPostC promoted autophagy against I/R injury at least partially via the activation of nNOS/AMPK/mTOR pathway.
Keywords: adenosine monophosphate-activated protein kinase (AMPK); autophagy; ischemia/reperfusion; ischemic postconditioning (IPostC); mammalian target of rapamycin (mTOR); neuronal nitric oxide synthase (nNOS).
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Ischemic postconditioning protects the heart against ischemia-reperfusion injury via neuronal nitric oxide synthase in the sarcoplasmic reticulum and mitochondria.Cell Death Dis. 2016 May 12;7(5):e2222. doi: 10.1038/cddis.2016.108. Cell Death Dis. 2016. PMID: 27171264 Free PMC article.
-
Effect of eNOS on Ischemic Postconditioning-Induced Autophagy against Ischemia/Reperfusion Injury in Mice.Biomed Res Int. 2019 Feb 10;2019:5201014. doi: 10.1155/2019/5201014. eCollection 2019. Biomed Res Int. 2019. PMID: 30881990 Free PMC article.
-
Ischemic postconditioning regulates cardiomyocyte autophagic activity following ischemia/reperfusion injury.Mol Med Rep. 2015 Jul;12(1):1169-76. doi: 10.3892/mmr.2015.3533. Epub 2015 Mar 24. Mol Med Rep. 2015. PMID: 25816157
-
Regulation of autophagy of the heart in ischemia and reperfusion.Apoptosis. 2023 Feb;28(1-2):55-80. doi: 10.1007/s10495-022-01786-1. Epub 2022 Nov 11. Apoptosis. 2023. PMID: 36369366 Review.
-
Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes.Oxid Med Cell Longev. 2017;2017:6437467. doi: 10.1155/2017/6437467. Epub 2017 Feb 19. Oxid Med Cell Longev. 2017. PMID: 28298952 Free PMC article. Review.
Cited by
-
Adaptive mechanisms in no flow vs. low flow ischemia in equine jejunum epithelium: Different paths to the same destination.Front Vet Sci. 2022 Sep 8;9:947482. doi: 10.3389/fvets.2022.947482. eCollection 2022. Front Vet Sci. 2022. PMID: 36157182 Free PMC article.
-
Ischemic Postconditioning-Mediated DJ-1 Activation Mitigate Intestinal Mucosa Injury Induced by Myocardial Ischemia Reperfusion in Rats Through Keap1/Nrf2 Pathway.Front Mol Biosci. 2021 Apr 30;8:655619. doi: 10.3389/fmolb.2021.655619. eCollection 2021. Front Mol Biosci. 2021. PMID: 33996908 Free PMC article.
-
Pioglitazone abrogates testicular damage induced by testicular torsion/detorsion in rats.Iran J Basic Med Sci. 2019 Aug;22(8):884-892. doi: 10.22038/ijbms.2019.33199.7929. Iran J Basic Med Sci. 2019. PMID: 31579444 Free PMC article.
-
Ischemic Postconditioning Alleviates Cerebral Ischemia-Reperfusion Injury Through Activating Autophagy During Early Reperfusion in Rats.Neurochem Res. 2018 Sep;43(9):1826-1840. doi: 10.1007/s11064-018-2599-3. Epub 2018 Jul 25. Neurochem Res. 2018. PMID: 30046966 Free PMC article.
-
Pharmacological postconditioning with Neuregulin-1 mimics the cardioprotective effects of ischaemic postconditioning via ErbB4-dependent activation of reperfusion injury salvage kinase pathway.Mol Med. 2018 Jul 31;24(1):39. doi: 10.1186/s10020-018-0040-7. Mol Med. 2018. PMID: 30134819 Free PMC article.
References
-
- Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Després J.P., Fullerton H.J., et al. Heart disease and stroke statistics-2016 update a report from the American Heart Association. Circulation. 2016;133:e38–e48. doi: 10.1161/CIR.0000000000000350. - DOI - PubMed
-
- Li F., Fan X., Zhang Y., Pang L., Ma X., Song M., Kou J., Yu B. Cardioprotection by combination of three compounds from ShengMai preparations in mice with myocardial ischemia/reperfusion injury through AMPK activation-mediated mitochondrial fission. Sci. Rep. 2016 doi: 10.1038/srep37114. - DOI - PMC - PubMed
-
- Guo J., Wang S.B., Yuan T.Y., Wu Y.J., Yan Y., Li L., Xu X.N., Gong L.L., Qin H.L., Fang L.H., et al. Coptisine protects rat heart against myocardial ischemia/reperfusion injury by suppressing myocardial apoptosis and inflammation. Atherosclerosis. 2013;231:384–391. doi: 10.1016/j.atherosclerosis.2013.10.003. - DOI - PubMed
-
- Shahzad T., Kasseckert S.A., Iraqi W., Johnson V., Schulz R., Schlüter K.D., Dörr O., Parahuleva M., Hamm C., Ladilov Y., et al. Mechanisms involved in postconditioning protection of cardiomyocytes against acute reperfusion injury. Mol. Cell. Cardiol. 2013;58:209–216. doi: 10.1016/j.yjmcc.2013.01.003. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
