

Multivariate simulation framework reveals performance of multi-trait GWAS methods
- PMID: 28287610
- PMCID: PMC5347376
- DOI: 10.1038/srep38837
Multivariate simulation framework reveals performance of multi-trait GWAS methods
Abstract
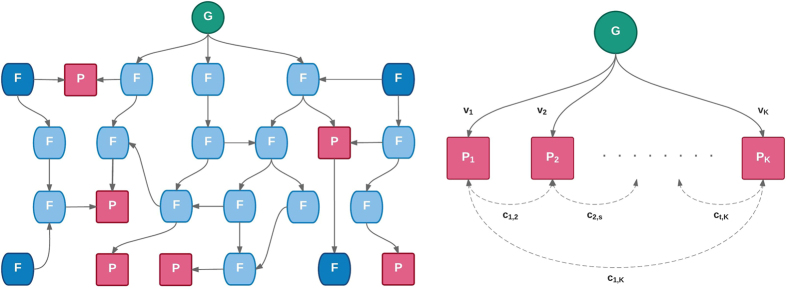
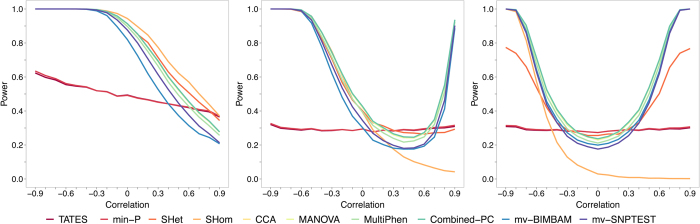
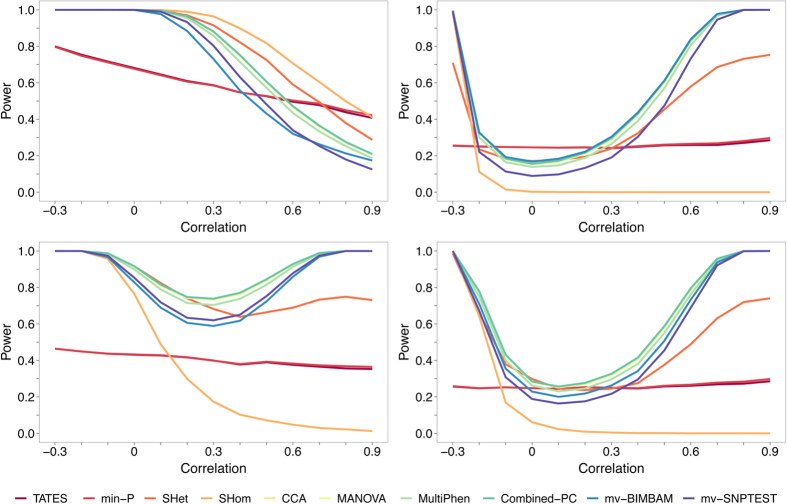
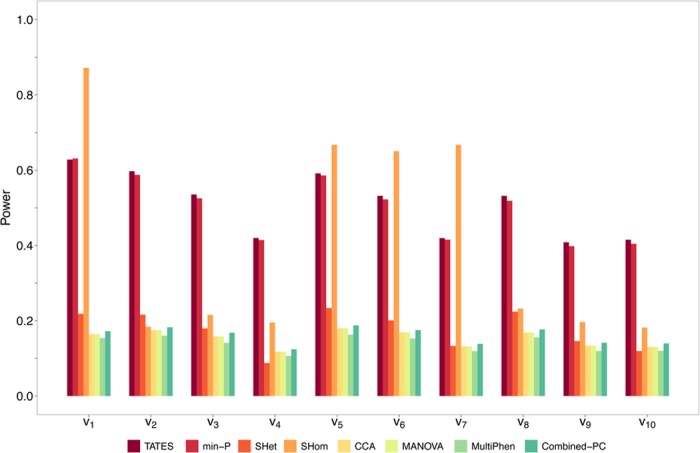
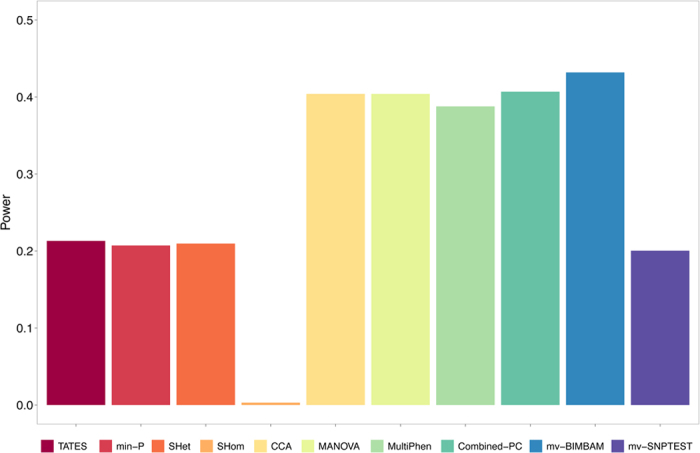
Burgeoning availability of genome-wide association study (GWAS) results and national biobank data has led to growing interest in performing multi-trait genetic analyses. Numerous multi-trait GWAS methods that exploit either summary statistics or individual-level data have been developed, but their relative performance is unclear. Here we develop a simulation framework to model the complex networks underlying multivariate genetic epidemiology, enabling the vast model space of genetic effects on multiple correlated traits to be explored systematically. We perform a comprehensive comparison of the leading multi-trait GWAS methods, finding: (1) method performance is highly sensitive to the specific combination of genetic effects and phenotypic correlations, (2) most of the current multivariate methods have remarkably similar statistical power, and (3) multivariate methods may offer a substantial increase in the discovery of genetic variants over the standard univariate approach. We believe our findings offer the clearest picture to date of the relative performance of multi-trait GWAS methods and act as a guide for method selection. We provide a web application and open-source software program implementing our simulation framework, for: (i) further benchmarking of multivariate GWAS methods, (ii) power calculations for multivariate genetic studies, and (iii) generating data for testing any multivariate method in genetic epidemiology.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
