

Determining Clostridium difficile intra-taxa diversity by mining multilocus sequence typing databases
- PMID: 28288567
- PMCID: PMC5348806
- DOI: 10.1186/s12866-017-0969-7
Determining Clostridium difficile intra-taxa diversity by mining multilocus sequence typing databases
Abstract
Background: Multilocus sequence typing (MLST) is a highly discriminatory typing strategy; it is reproducible and scalable. There is a MLST scheme for Clostridium difficile (CD), a gram positive bacillus causing different pathologies of the gastrointestinal tract. This work was aimed at describing the frequency of sequence types (STs) and Clades (C) reported and evalute the intra-taxa diversity in the CD MLST database (CD-MLST-db) using an MLSA approach.
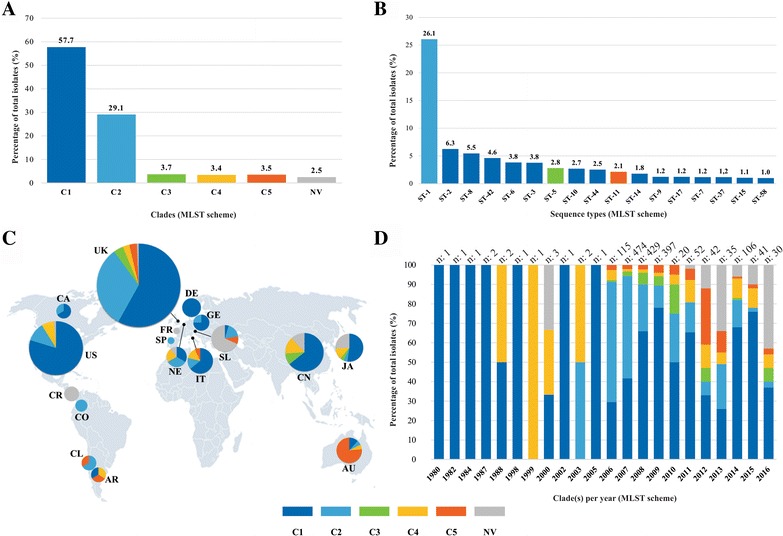
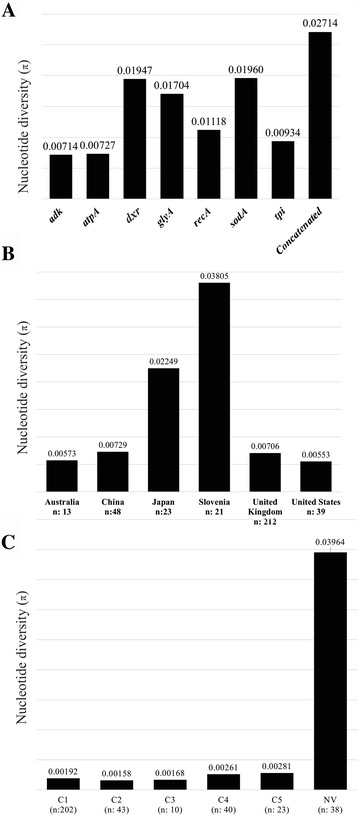
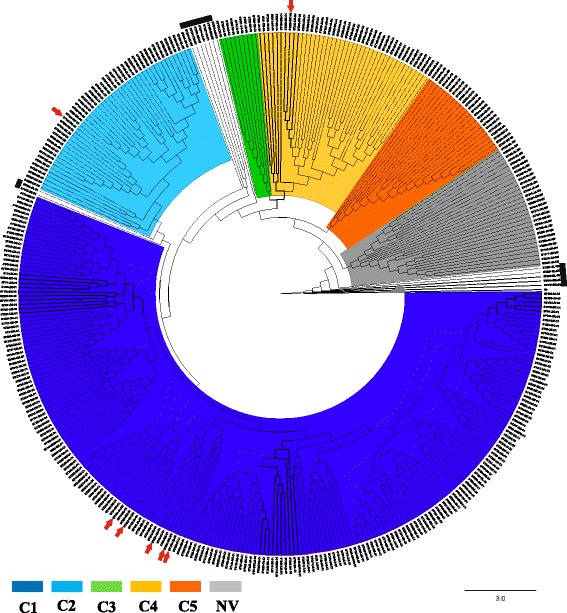
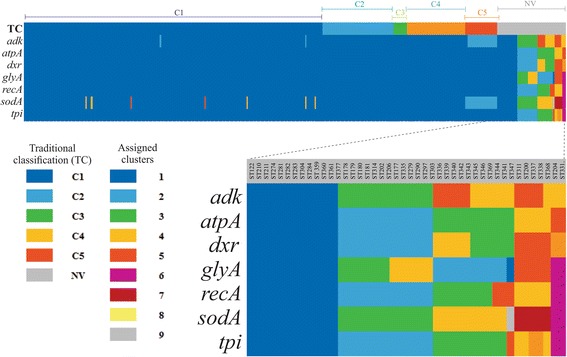
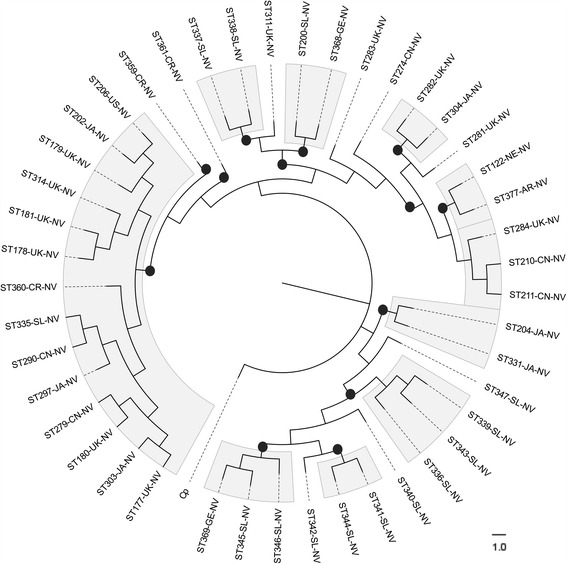
Results: Analysis of 1778 available isolates showed that clade 1 (C1) was the most frequent worldwide (57.7%), followed by C2 (29.1%). Regarding sequence types (STs), it was found that ST-1, belonging to C2, was the most frequent. The isolates analysed came from 17 countries, mostly from the United Kingdom (UK) (1541 STs, 87.0%). The diversity of the seven housekeeping genes in the MLST scheme was evaluated, and alleles from the profiles (STs), for identifying CD population structure. It was found that adk and atpA are conserved genes allowing a limited amount of clusters to be discriminated; however, different genes such as drx, glyA and particularly sodA showed high diversity indexes and grouped CD populations in many clusters, suggesting that these genes' contribution to CD typing should be revised. It was identified that CD STs reported to date have a mostly clonal population structure with foreseen events of recombination; however, one group of STs was not assigned to a clade being highly different containing at least nine well-supported clusters, suggesting a greater amount of clades for CD.
Conclusions: This study shows the usefulness of CD-MLST-db as a tool for studying CD distribution and population structure, identifying the need for reviewing the usefulness of sodA as housekeeping gene within the MLST scheme and suggesting the existence of a greater amount of CD clades. The study also shows the plausible exchange of genetic material between STs, contributing towards intra-taxa genetic diversity.
Keywords: Clostridium difficile; Distribution pattern; Multilocus sequence typing (MLST); Population structure.
Figures
Similar articles
-
Multilocus sequence typing of Clostridium difficile.J Clin Microbiol. 2010 Mar;48(3):770-8. doi: 10.1128/JCM.01796-09. Epub 2009 Dec 30. J Clin Microbiol. 2010. PMID: 20042623 Free PMC article.
-
Comparative analysis of an expanded Clostridium difficile reference strain collection reveals genetic diversity and evolution through six lineages.Infect Genet Evol. 2012 Oct;12(7):1577-85. doi: 10.1016/j.meegid.2012.06.003. Epub 2012 Jun 15. Infect Genet Evol. 2012. PMID: 22705462
-
Multilocus variable-number tandem-repeat analysis and multilocus sequence typing reveal genetic relationships among Clostridium difficile isolates genotyped by restriction endonuclease analysis.J Clin Microbiol. 2010 Feb;48(2):412-8. doi: 10.1128/JCM.01315-09. Epub 2009 Dec 2. J Clin Microbiol. 2010. PMID: 19955268 Free PMC article.
-
Microbiological features, epidemiology, and clinical presentation of Clostridioidesdifficile strains from MLST Clade 2: A narrative review.Anaerobe. 2021 Jun;69:102355. doi: 10.1016/j.anaerobe.2021.102355. Epub 2021 Mar 10. Anaerobe. 2021. PMID: 33711422 Review.
-
Genomic diversity of Clostridium difficile strains.Res Microbiol. 2015 May;166(4):353-60. doi: 10.1016/j.resmic.2015.02.002. Epub 2015 Feb 18. Res Microbiol. 2015. PMID: 25700631 Review.
Cited by
-
New gene markers for classification and quantification of Faecalibacterium spp. in the human gut.FEMS Microbiol Ecol. 2023 Apr 7;99(5):fiad035. doi: 10.1093/femsec/fiad035. FEMS Microbiol Ecol. 2023. PMID: 36990641 Free PMC article.
-
Molecular Epidemiology and Risk Factors of Clostridium difficile ST81 Infection in a Teaching Hospital in Eastern China.Front Cell Infect Microbiol. 2020 Dec 23;10:578098. doi: 10.3389/fcimb.2020.578098. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 33425775 Free PMC article.
-
Community-acquired infection with hypervirulent Clostridium difficile isolates that carry different toxin and antibiotic resistance loci: a case report.Gut Pathog. 2017 Nov 9;9:63. doi: 10.1186/s13099-017-0212-y. eCollection 2017. Gut Pathog. 2017. PMID: 29151897 Free PMC article.
-
Whole genome analysis reveals new insights into the molecular characteristics of Clostridioides difficile NAP1/BI/027/ST1 clinical isolates in the People's Republic of China.Infect Drug Resist. 2019 Jul 1;12:1783-1794. doi: 10.2147/IDR.S203238. eCollection 2019. Infect Drug Resist. 2019. PMID: 31308704 Free PMC article.
-
Unveiling the Multilocus Sequence Typing (MLST) Schemes and Core Genome Phylogenies for Genotyping Chlamydia trachomatis.Front Microbiol. 2018 Aug 22;9:1854. doi: 10.3389/fmicb.2018.01854. eCollection 2018. Front Microbiol. 2018. PMID: 30186244 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
