

Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening
- PMID: 28288978
- PMCID: PMC5524036
- DOI: 10.1161/JAHA.116.005328
Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening
Abstract
Background: The cardiac microvascular system ischemia/reperfusion injury following percutaneous coronary intervention is a clinical thorny problem. This study explores the mechanisms by which ischemia/reperfusion injury induces cardiac microcirculation collapse.
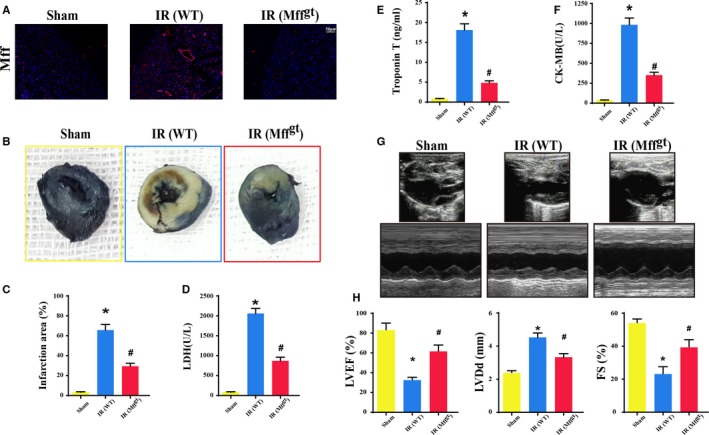
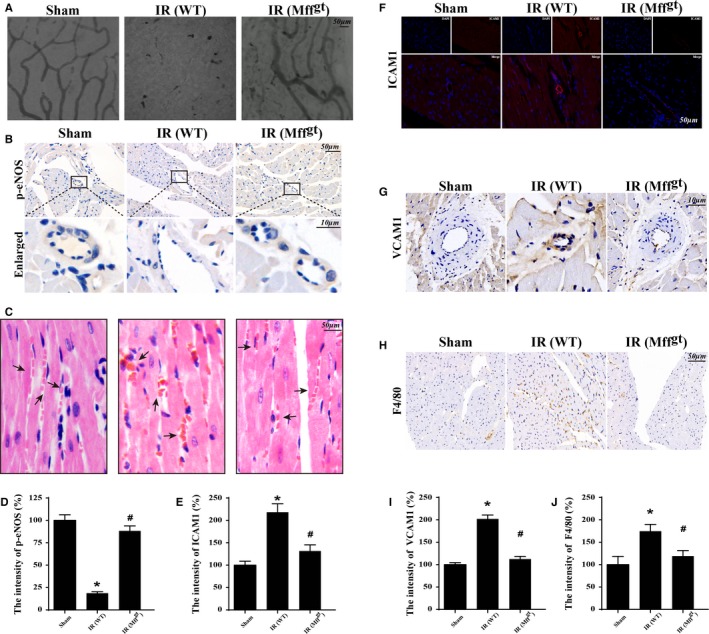
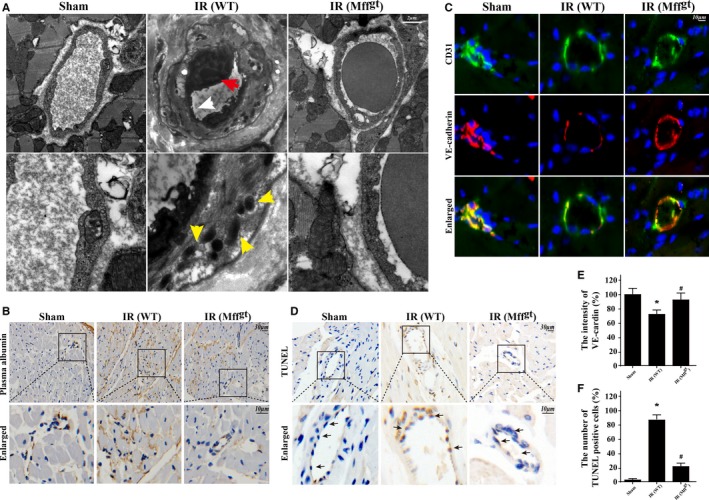
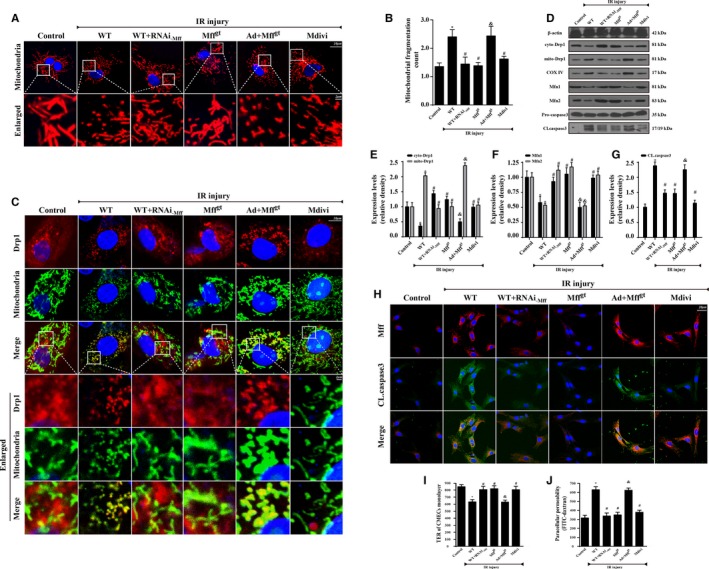
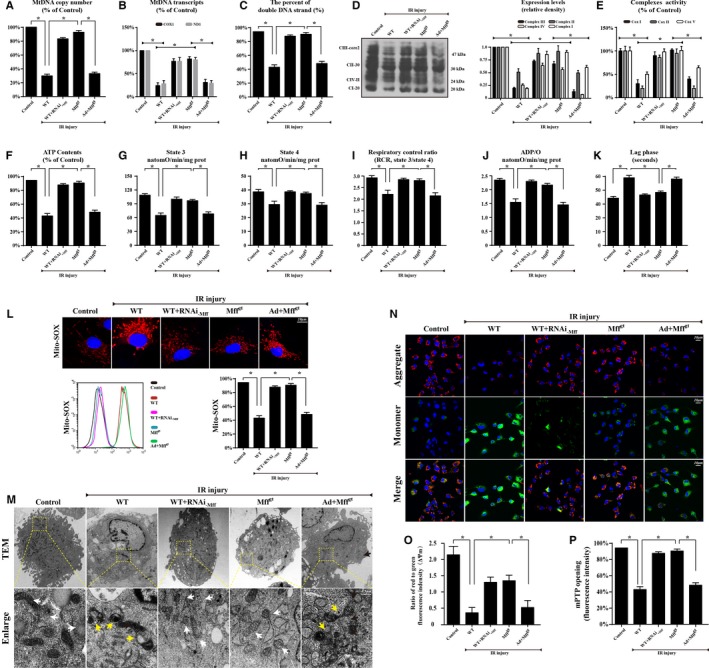
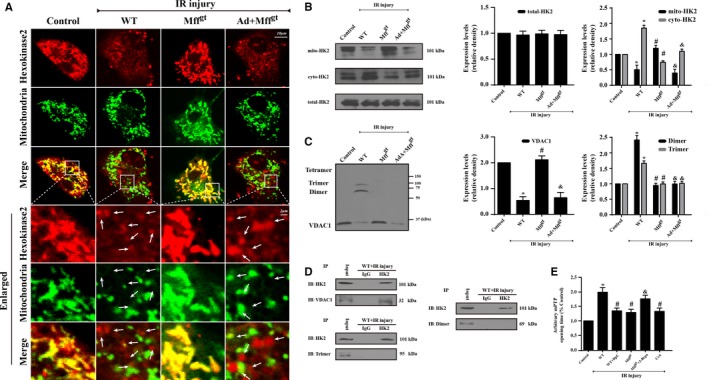
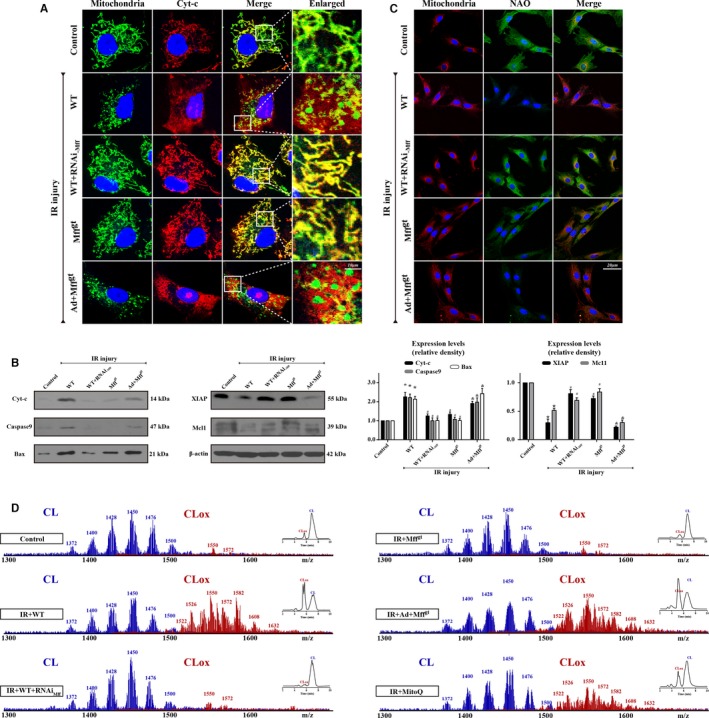
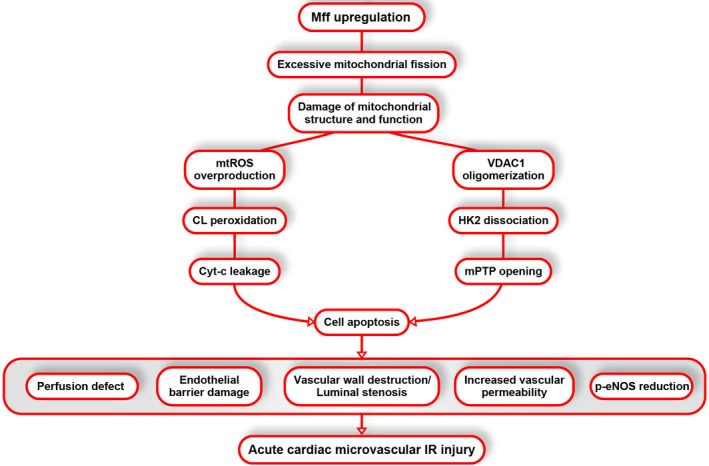
Methods and results: In wild-type mice, mitochondrial fission factor (Mff) expression increased in response to acute microvascular ischemia/reperfusion injury. Compared with wild-type mice, homozygous Mff-deficient (Mffgt) mice exhibited a smaller infarcted area, restored cardiac function, improved blood flow, and reduced microcirculation perfusion defects. Histopathology analysis demonstrated that cardiac microcirculation endothelial cells (CMECs) in Mffgt mice had an intact endothelial barrier, recovered phospho-endothelial nitric oxide synthase production, opened lumen, undivided mitochondrial structures, and less CMEC death. In vitro, Mff-deficient CMECs (derived from Mffgt mice or Mff small interfering RNA-treated) demonstrated less mitochondrial fission and mitochondrial-dependent apoptosis compared with cells derived from wild-type mice. The loss of Mff inhibited mitochondrial permeability transition pore opening via blocking the oligomerization of voltage-dependent anion channel 1 and subsequent hexokinase 2 separation from mitochondria. Moreover, Mff deficiency reduced the cyt-c leakage into the cytoplasm by alleviating cardiolipin oxidation resulting from damage to the electron transport chain complexes and mitochondrial reactive oxygen species overproduction.
Conclusions: This evidence clearly illustrates that microcirculatory ischemia/reperfusion injury can be attributed to Mff-dependent mitochondrial fission via voltage-dependent anion channel 1/hexokinase 2-mediated mitochondrial permeability transition pore opening and mitochondrial reactive oxygen species/cardiolipin involved cyt-c release.
Keywords: apoptosis; endothelial cell; ischemia/reperfusion injury; mitochondria.
© 2017 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures
References
-
- Dauber IM, VanBenthuysen KM, McMurtry IF, Wheeler GS, Lesnefsky EJ, Horwitz LD, Weil JV. Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ Res. 1990;66:986–998. - PubMed
-
- Ito H. No‐reflow phenomenon and prognosis in patients with acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2006;3:499–506. - PubMed
-
- Weir RA, Murphy CA, Petrie CJ, Martin TN, Balmain S, Clements S, Steedman T, Wagner GS, Dargie HJ, McMurray JJ. Microvascular obstruction remains a portent of adverse remodeling in optimally treated patients with left ventricular systolic dysfunction after acute myocardial infarction. Circ Cardiovasc Imaging. 2010;3:360–367. - PubMed
-
- Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013;75:95–126. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
