

Renal Tubular Ubiquitin-Protein Ligase NEDD4-2 Is Required for Renal Adaptation during Long-Term Potassium Depletion
- PMID: 28289184
- PMCID: PMC5533229
- DOI: 10.1681/ASN.2016070732
Renal Tubular Ubiquitin-Protein Ligase NEDD4-2 Is Required for Renal Adaptation during Long-Term Potassium Depletion
Abstract
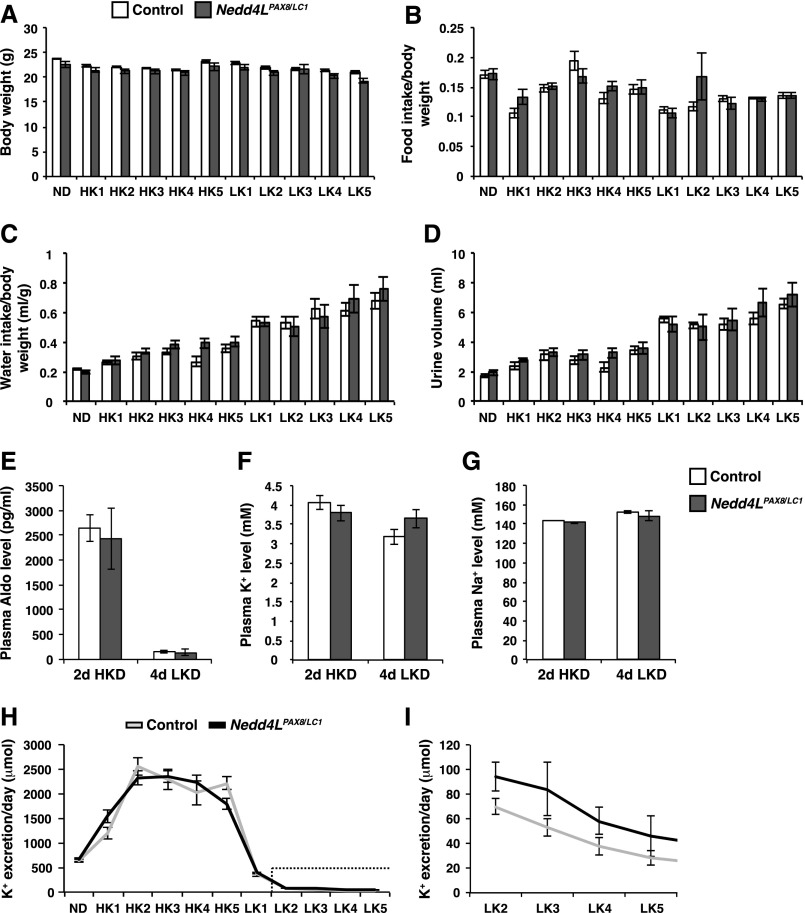
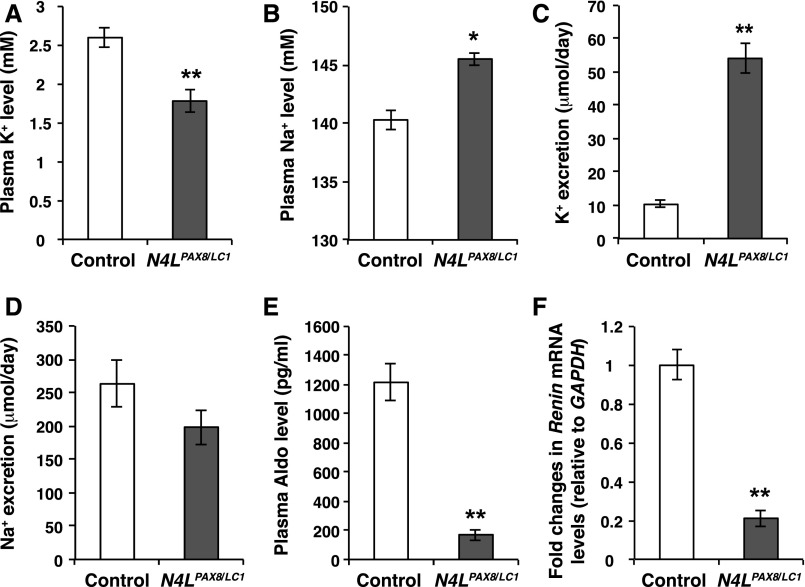
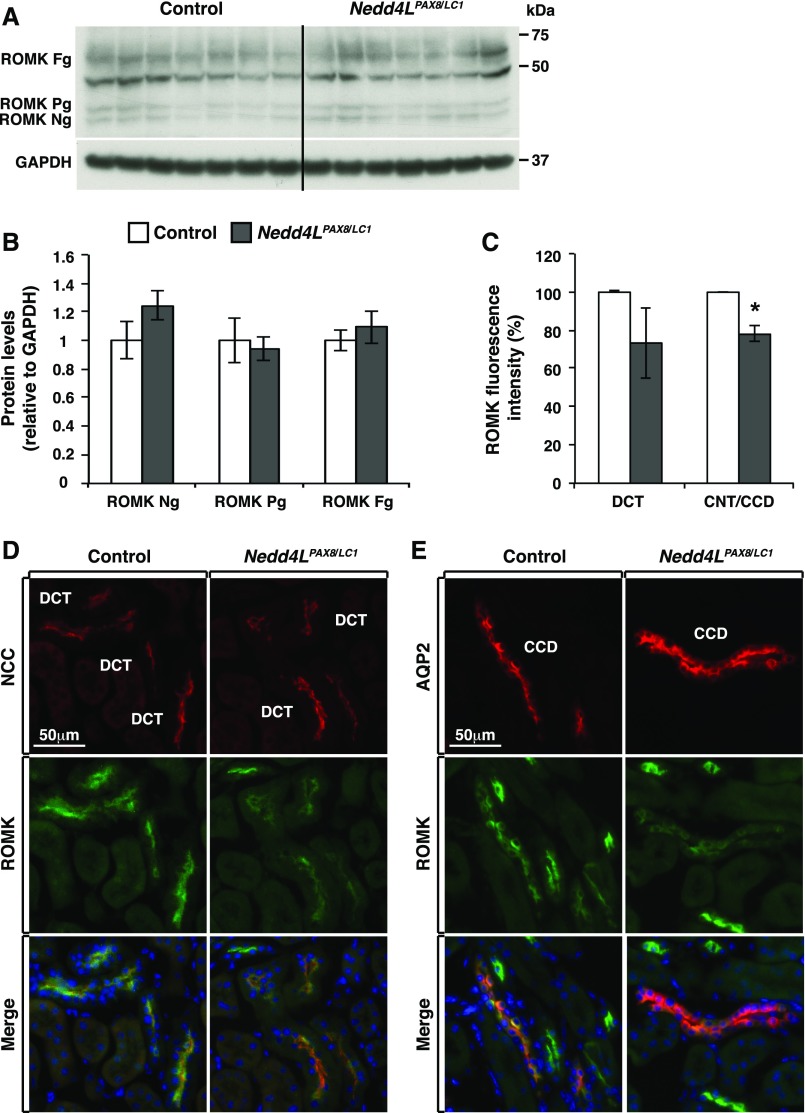
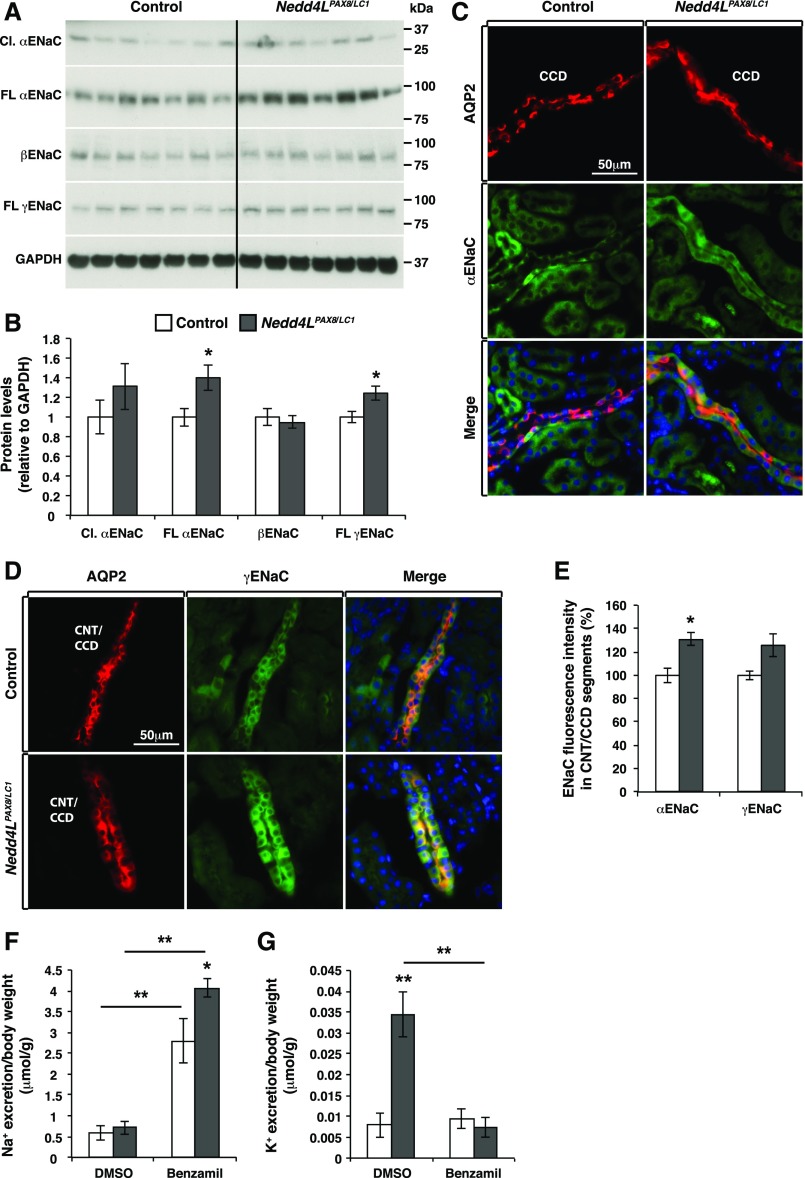
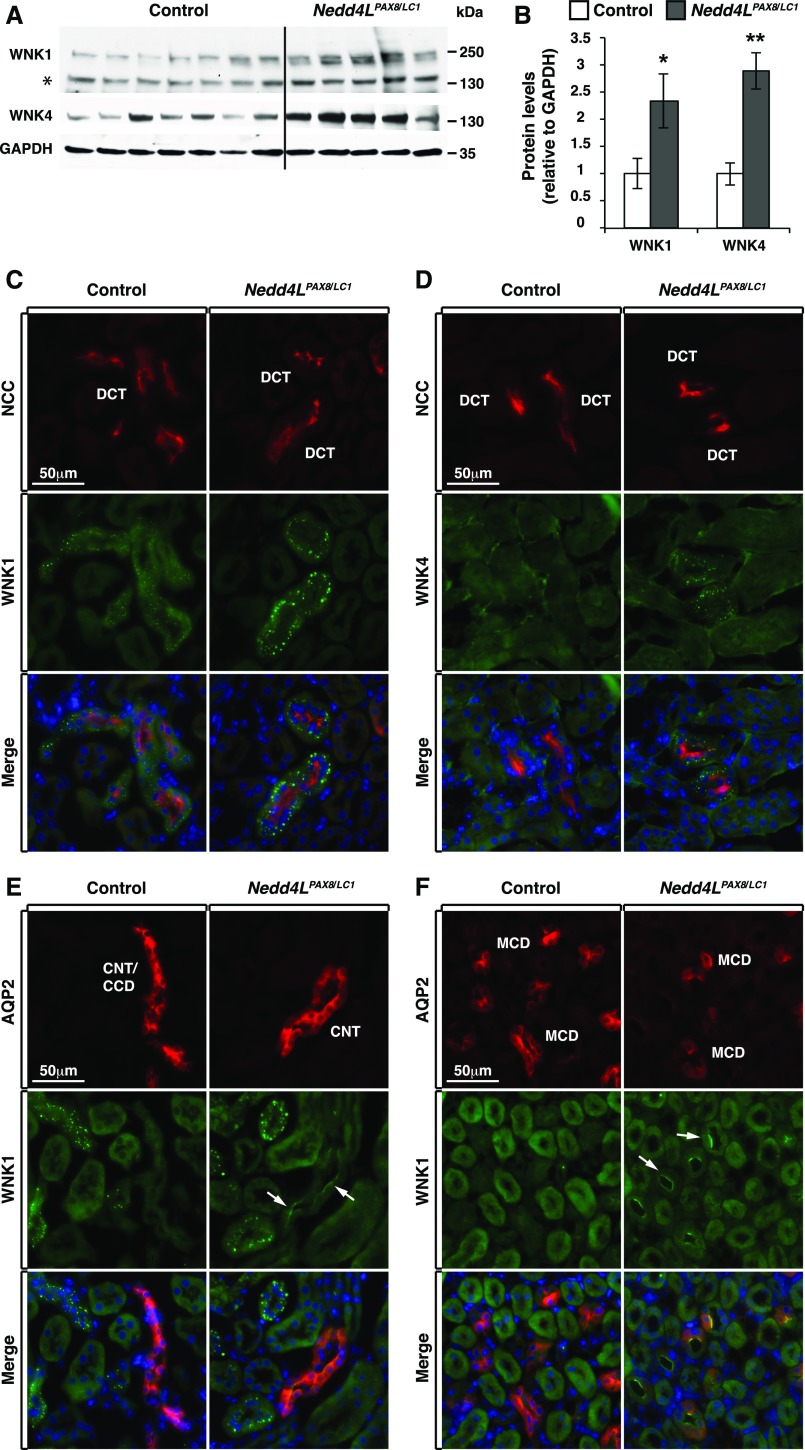
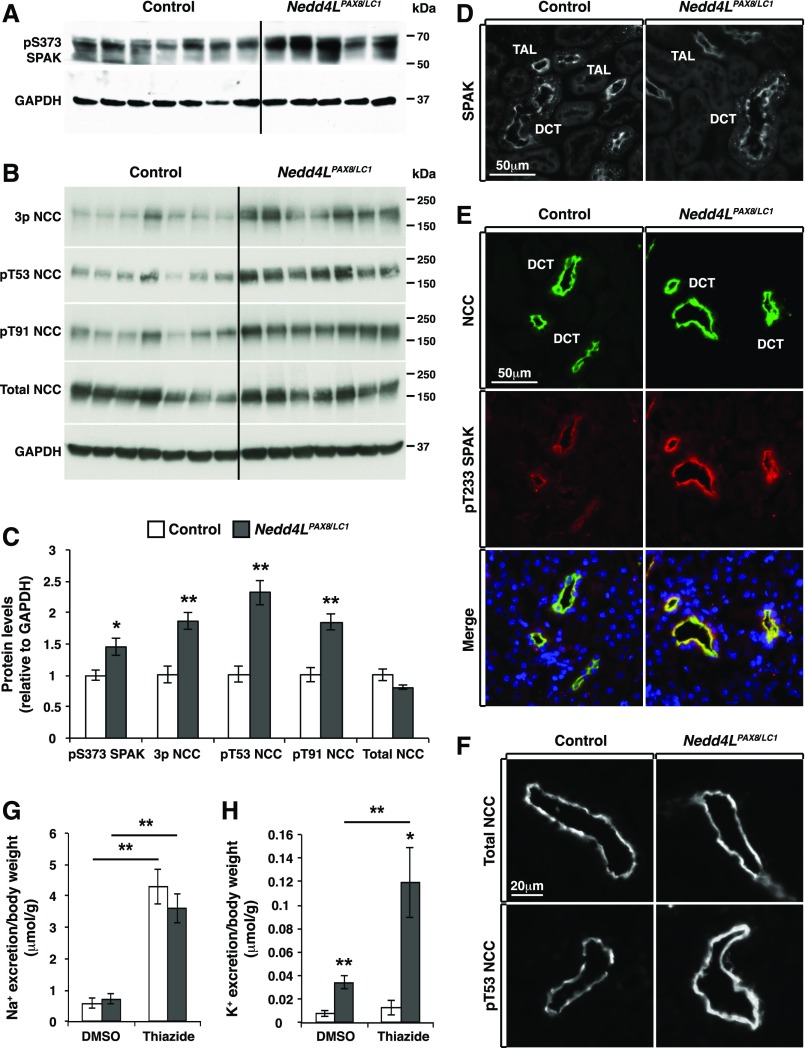
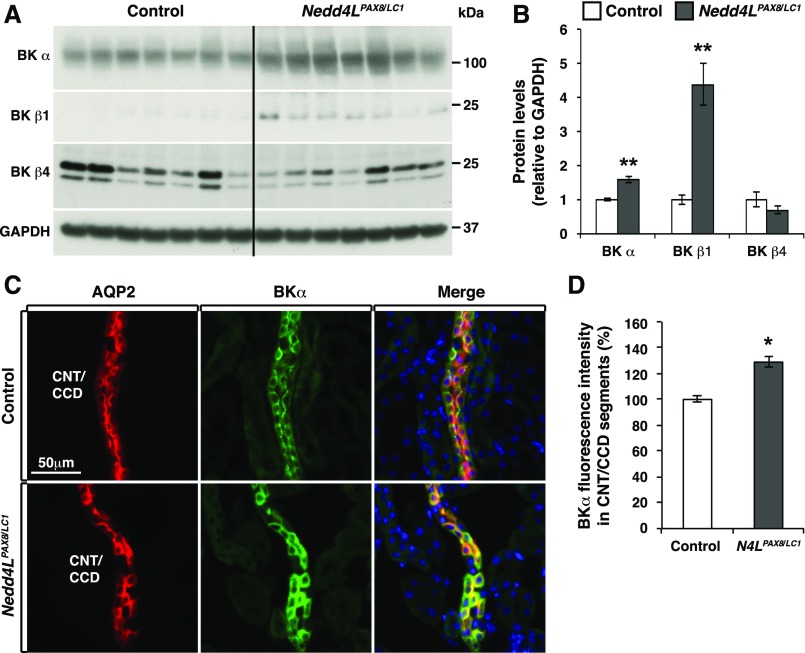
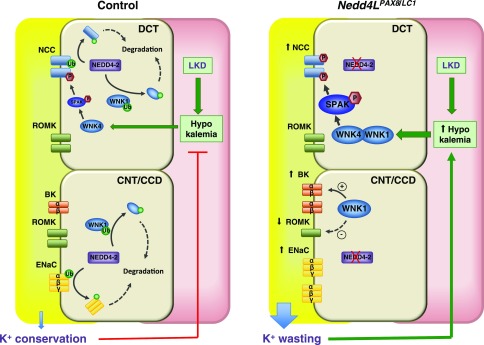
Adaptation of the organism to potassium (K+) deficiency requires precise coordination among organs involved in K+ homeostasis, including muscle, liver, and kidney. How the latter performs functional and molecular changes to ensure K+ retention is not well understood. Here, we investigated the role of ubiquitin-protein ligase NEDD4-2, which negatively regulates the epithelial sodium channel (ENaC), Na+/Cl- cotransporter (NCC), and with no-lysine-kinase 1 (WNK1). After dietary K+ restriction for 2 weeks, compared with control littermates, inducible renal tubular NEDD4-2 knockout (Nedd4LPax8/LC1 ) mice exhibited severe hypokalemia and urinary K+ wasting. Notably, expression of the ROMK K+ channel did not change in the distal convoluted tubule and decreased slightly in the cortical/medullary collecting duct, whereas BK channel abundance increased in principal cells of the connecting tubule/collecting ducts. However, K+ restriction also enhanced ENaC expression in Nedd4LPax8/LC1 mice, and treatment with the ENaC inhibitor, benzamil, reversed excessive K+ wasting. Moreover, K+ restriction increased WNK1 and WNK4 expression and enhanced SPAK-mediated NCC phosphorylation in Nedd4LPax8/LC1 mice, with no change in total NCC. We propose a mechanism in which NEDD4-2 deficiency exacerbates hypokalemia during dietary K+ restriction primarily through direct upregulation of ENaC, whereas increased BK channel expression has a less significant role. These changes outweigh the compensatory antikaliuretic effects of diminished ROMK expression, increased NCC phosphorylation, and enhanced WNK pathway activity in the distal convoluted tubule. Thus, NEDD4-2 has a crucial role in K+ conservation through direct and indirect effects on ENaC, distal nephron K+ channels, and WNK signaling.
Keywords: ENaC; K channels; ion transport; signaling.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
