

Humoral Immunodeficiency with Hypotonia, Feeding Difficulties, Enteropathy, and Mild Eczema Caused by a Classical FOXP3 Mutation
- PMID: 28289675
- PMCID: PMC5326763
- DOI: 10.3389/fped.2017.00037
Humoral Immunodeficiency with Hypotonia, Feeding Difficulties, Enteropathy, and Mild Eczema Caused by a Classical FOXP3 Mutation
Abstract
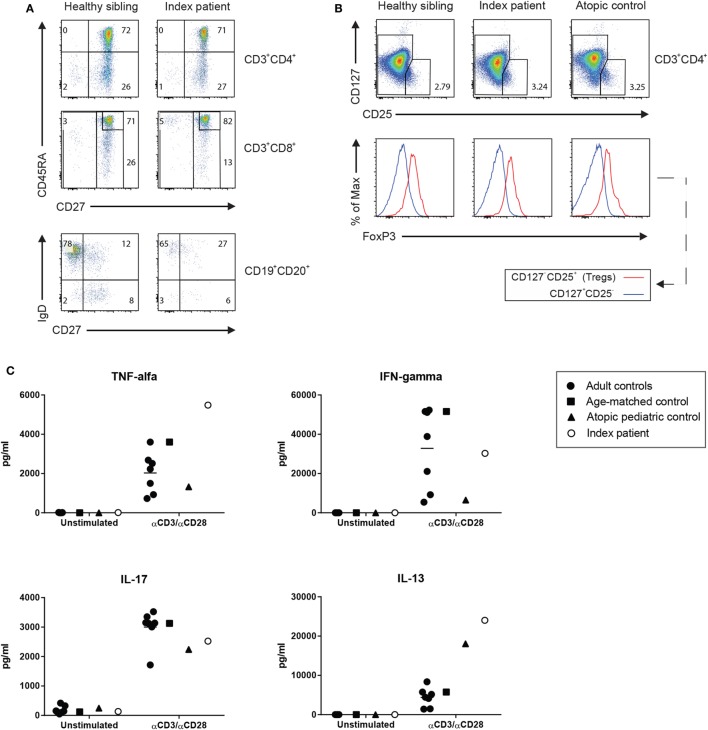
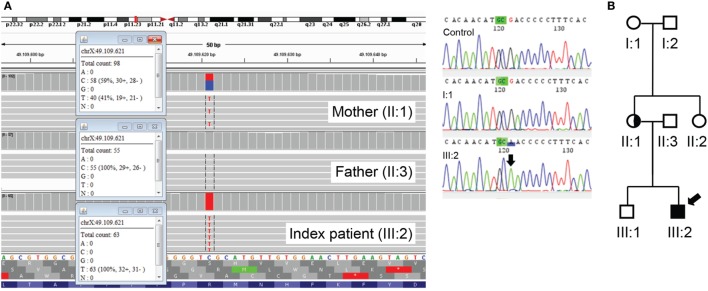
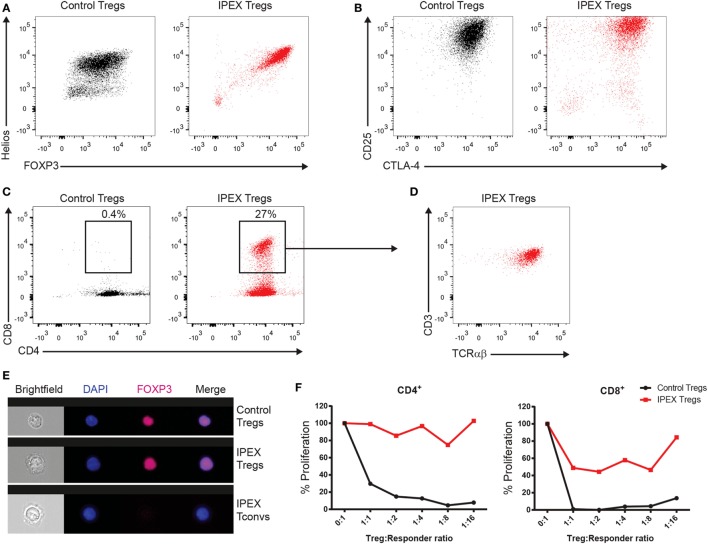
We describe here the case of a boy who presented with pulmonary infections, feeding difficulties due to velopharyngeal insufficiency and gastroesophageal reflux, myopathy, and hypotonia soon after birth. Later, he was also found to have an elevated immunoglobulin (Ig) E and mild eczema and was diagnosed with inflammatory bowel disease. Further immunological screening at the age of 7 years showed low B and NK cell numbers but normal CD4+ and CD8+ T cells and notably, normal numbers of CD4+ regulatory T (Treg) cells. Serum IgG, IgA, and IgM were low to normal, but he had a deficient response to a pneumococcal polysaccharide vaccine and thus a humoral immunodeficiency. To our surprise, whole exome sequencing revealed a mutation in forkhead box protein 3 (FOXP3), encoding an essential transcription factor for the development and function of Treg cells. This classical mutation is associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Further in vitro studies indeed showed defective function of Treg cells despite normal FOXP3 protein expression and nuclear localization. The boy underwent hematopoietic stem cell transplantation at 11 years of age and despite the temporary development of diabetes while on prednisone is now doing much better, IgE levels have declined, and his fatigue has improved. This case illustrates that a classical pathogenic mutation in FOXP3 can lead to a clinical phenotype where the diagnosis of IPEX syndrome was never considered because of the lack of diabetes and the presence of only mild eczema, in addition to the normal Treg cell numbers and FOXP3 expression.
Keywords: FOXP3; IPEX syndrome; Treg; WES; autoimmunity; immunodeficiency.
Figures
References
-
- Savova R, Arshinkova M, Houghton J, Konstantinova M, Gaydarova M, Georgieva E, et al. Clinical case of immune dysregulation, polyendocrinopaty, enteropathy, X-linked (IPEX) syndrome with severe immune deficiency and late onset of endocrinopathy and enteropathy. Case Rep Med (2014) 2014:564926. 10.1155/2014/564926 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous