

Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy
- PMID: 28289718
- PMCID: PMC5333962
- DOI: 10.1172/jci.insight.91225
Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy
Abstract
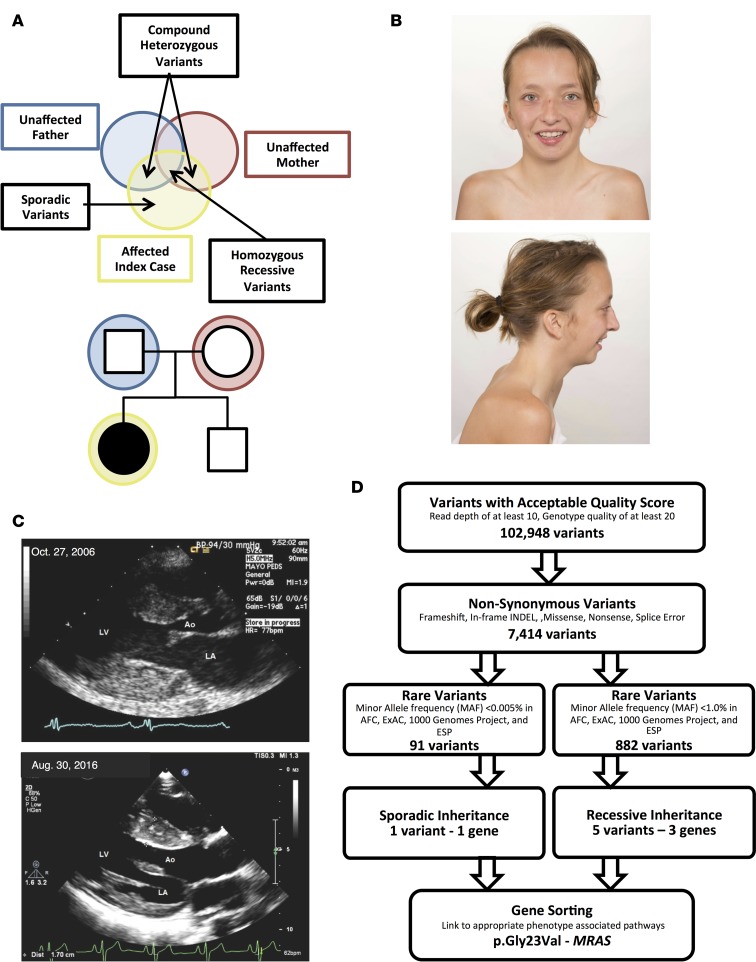
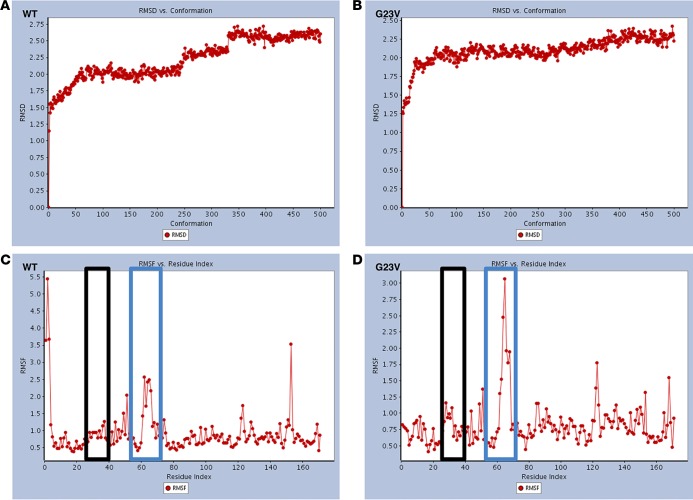
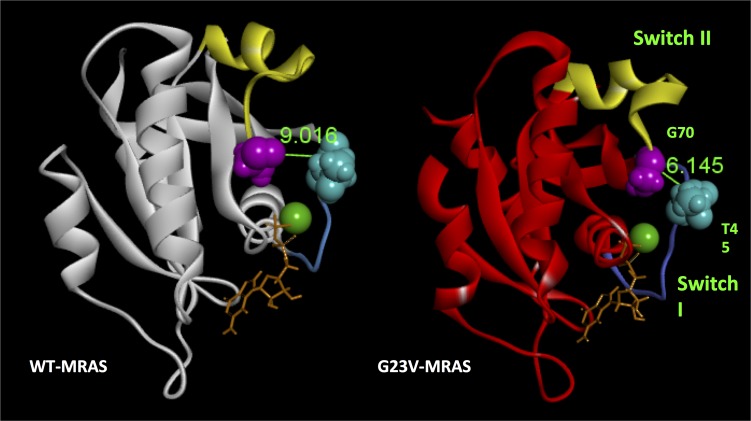
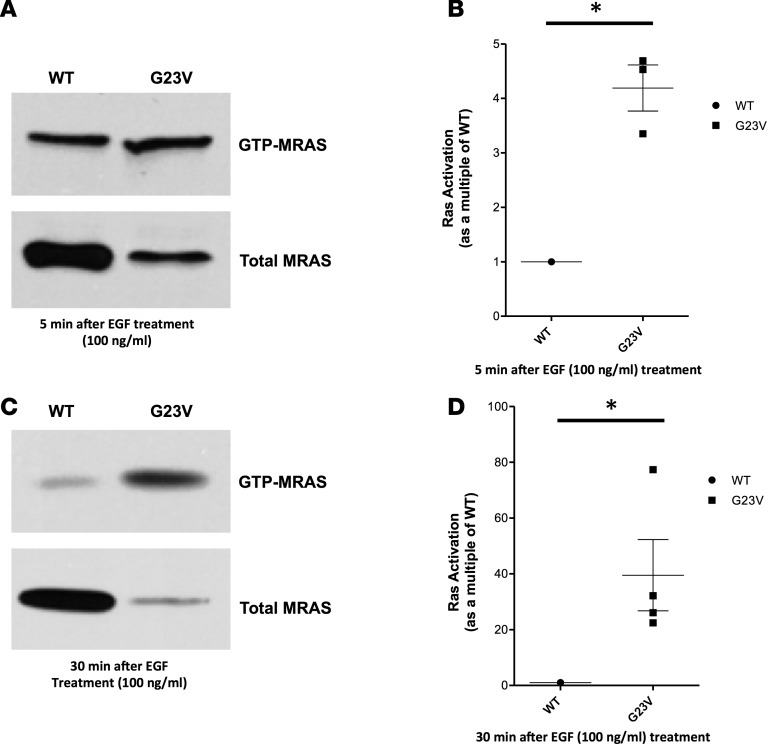
Noonan syndrome (NS; MIM 163950) is an autosomal dominant disorder and a member of a family of developmental disorders termed "RASopathies," which are caused mainly by gain-of-function mutations in genes encoding RAS/MAPK signaling pathway proteins. Whole exome sequencing (WES) and trio-based genomic triangulation of a 15-year-old female with a clinical diagnosis of NS and concomitant cardiac hypertrophy and her unaffected parents identified a de novo variant in MRAS-encoded RAS-related protein 3 as the cause of her disease. Mutation analysis using in silico mutation prediction tools and molecular dynamics simulations predicted the identified variant, p.Gly23Val-MRAS, to be damaging to normal protein function and adversely affect effector interaction regions and the GTP-binding site. Subsequent ectopic expression experiments revealed a 40-fold increase in MRAS activation for p.Gly23Val-MRAS compared with WT-MRAS. Additional biochemical assays demonstrated enhanced activation of both RAS/MAPK pathway signaling and downstream gene expression in cells expressing p.Gly23Val-MRAS. Mutational analysis of MRAS in a cohort of 109 unrelated patients with phenotype-positive/genotype-negative NS and cardiac hypertrophy yielded another patient with a sporadic de novo MRAS variant (p.Thr68Ile, c.203C>T). Herein, we describe the discovery of mutations in MRAS in patients with NS and cardiac hypertrophy, establishing MRAS as the newest NS with cardiac hypertrophy-susceptibility gene.
Conflict of interest statement
Conflict of interest: D.J. Tester and M.J. Ackerman receive royalties from Transgenomic for the FAMILION genetic tests. M.J. Ackerman is a consultant for or is on the advisory board of Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia, and St. Jude Medical.
Figures

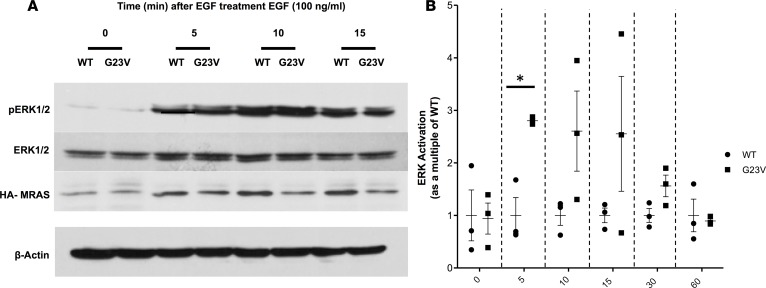
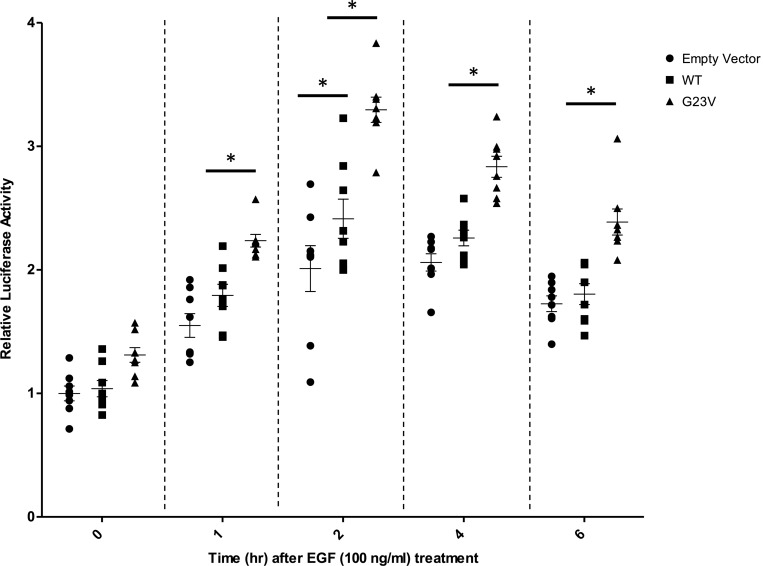
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
