

Integrated Molecular Characterization of Uterine Carcinosarcoma
- PMID: 28292439
- PMCID: PMC5599133
- DOI: 10.1016/j.ccell.2017.02.010
Integrated Molecular Characterization of Uterine Carcinosarcoma
Abstract
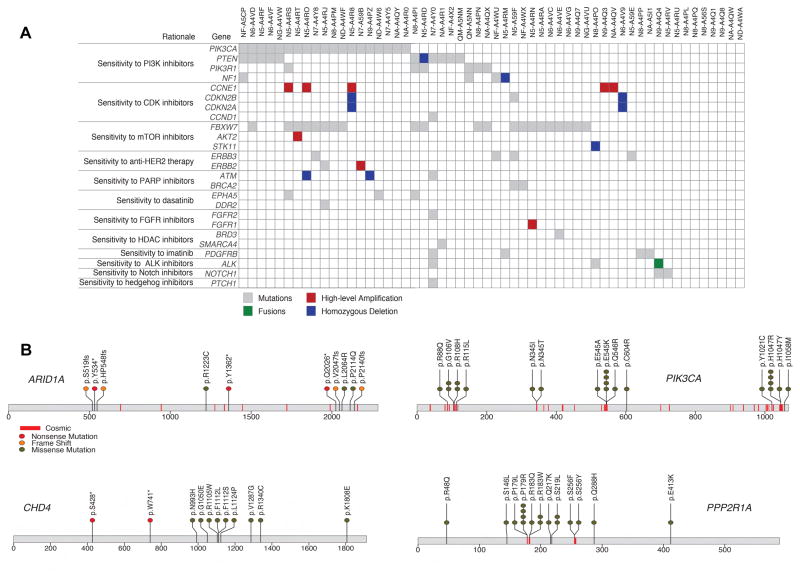
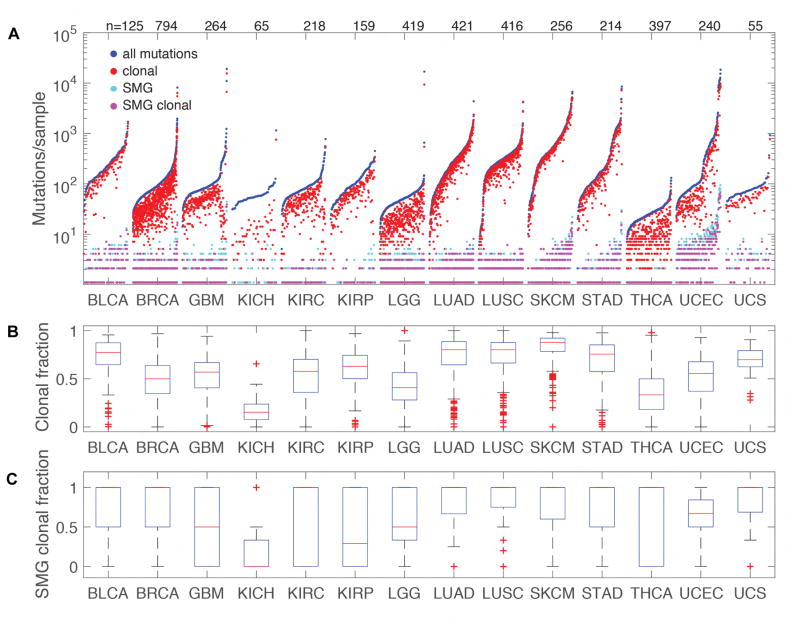
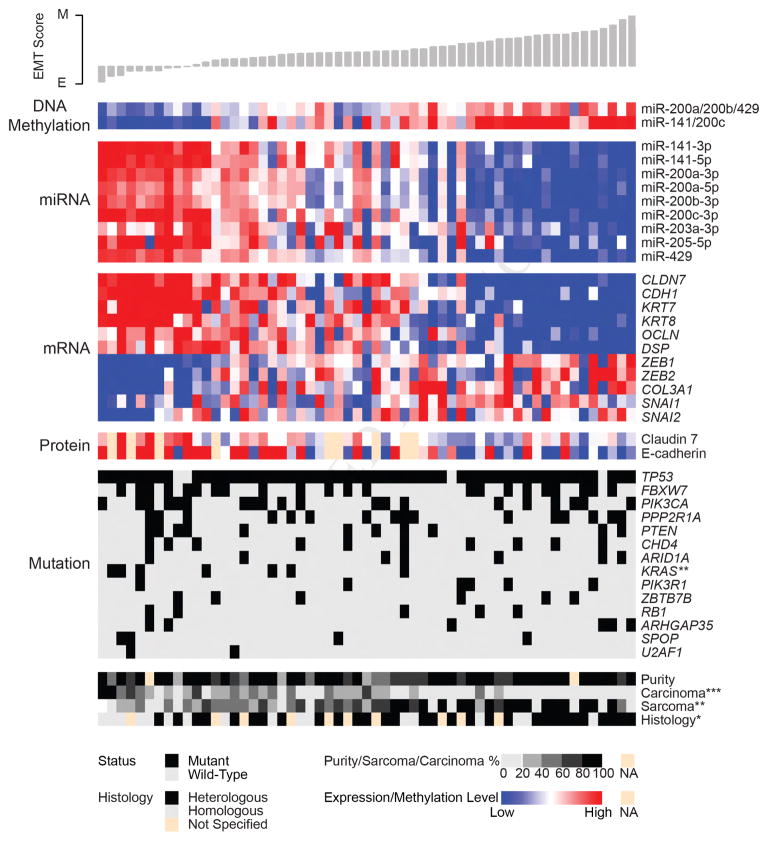
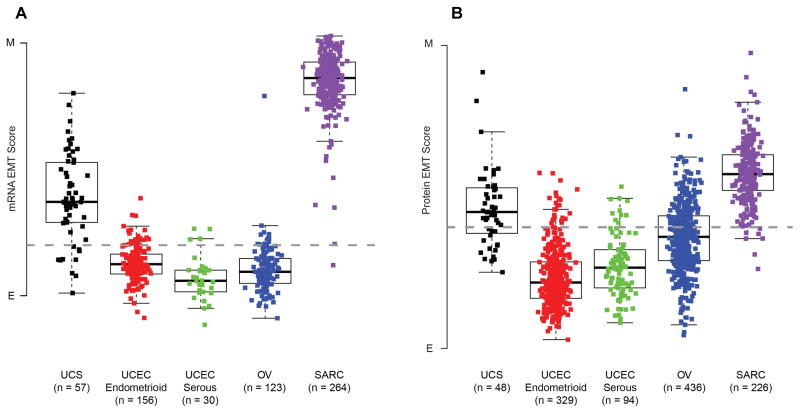
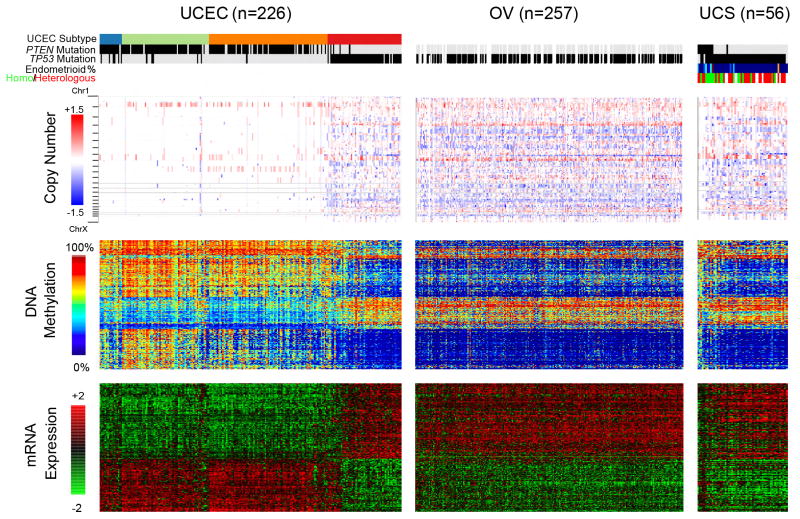
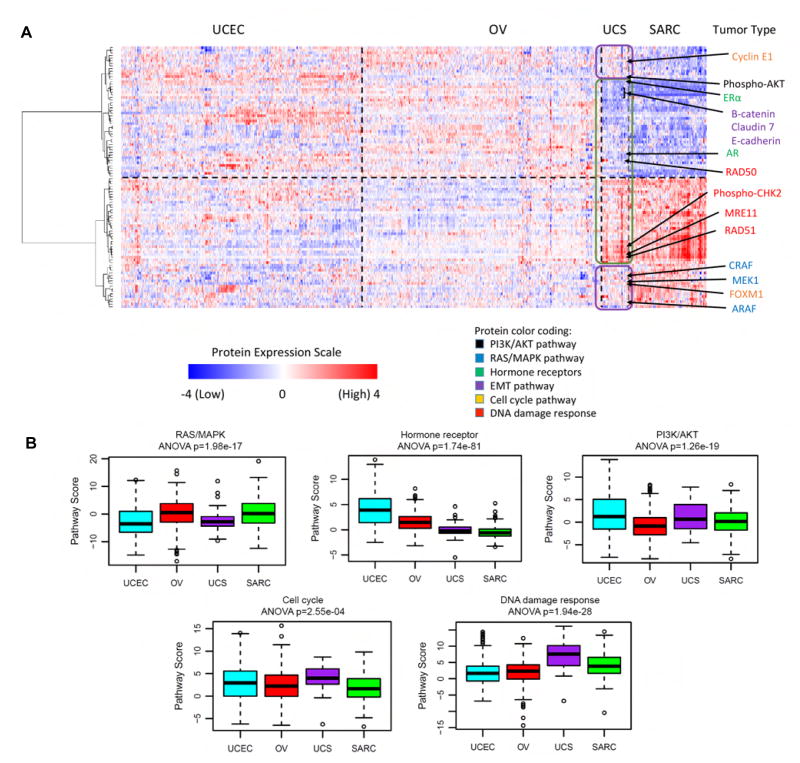
We performed genomic, epigenomic, transcriptomic, and proteomic characterizations of uterine carcinosarcomas (UCSs). Cohort samples had extensive copy-number alterations and highly recurrent somatic mutations. Frequent mutations were found in TP53, PTEN, PIK3CA, PPP2R1A, FBXW7, and KRAS, similar to endometrioid and serous uterine carcinomas. Transcriptome sequencing identified a strong epithelial-to-mesenchymal transition (EMT) gene signature in a subset of cases that was attributable to epigenetic alterations at microRNA promoters. The range of EMT scores in UCS was the largest among all tumor types studied via The Cancer Genome Atlas. UCSs shared proteomic features with gynecologic carcinomas and sarcomas with intermediate EMT features. Multiple somatic mutations and copy-number alterations in genes that are therapeutic targets were identified.
Keywords: EMT; TGGA; The Cancer Genome Atlas; UCS; endometrial cancer; epithelial-to-mesenchymal transition; gynecologic cancer; gynecologic oncology; translational science; uterine carcinosarcoma.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures

References
-
- Abeln EC, Smit VT, Wessels JW, de Leeuw WJ, Cornelisse CJ, Fleuren GJ. Molecular genetic evidence for the conversion hypothesis of the origin of malignant mixed mullerian tumours. The Journal of pathology. 1997;183:424–431. - PubMed
-
- Bland AE, Stone R, Heuser C, Shu J, Jazaeri A, Shutter J, Atkins K, Rice L. A clinical and biological comparison between malignant mixed mullerian tumors and grade 3 endometrioid endometrial carcinomas. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society. 2009;19:261–265. - PubMed
Publication types
MeSH terms
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA180951/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
