

Mechanisms of Melatonin in Alleviating Alzheimer's Disease
- PMID: 28294066
- PMCID: PMC5652010
- DOI: 10.2174/1570159X15666170313123454
Mechanisms of Melatonin in Alleviating Alzheimer's Disease
Abstract
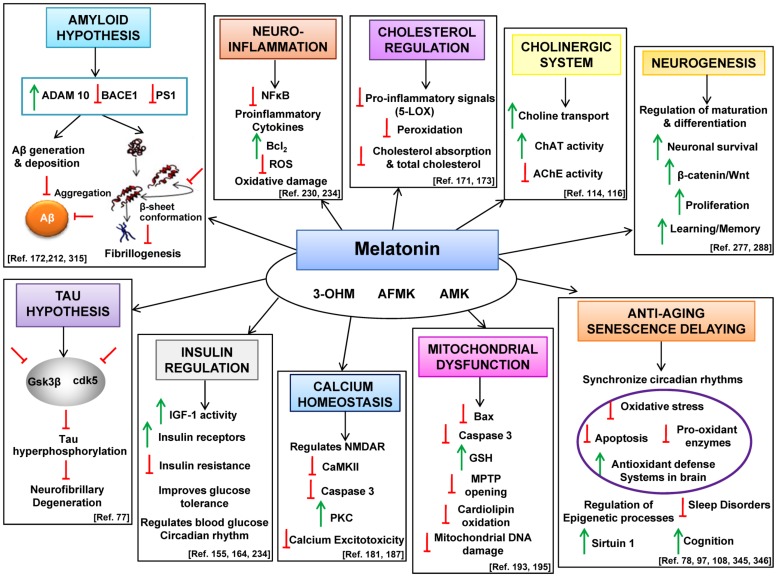
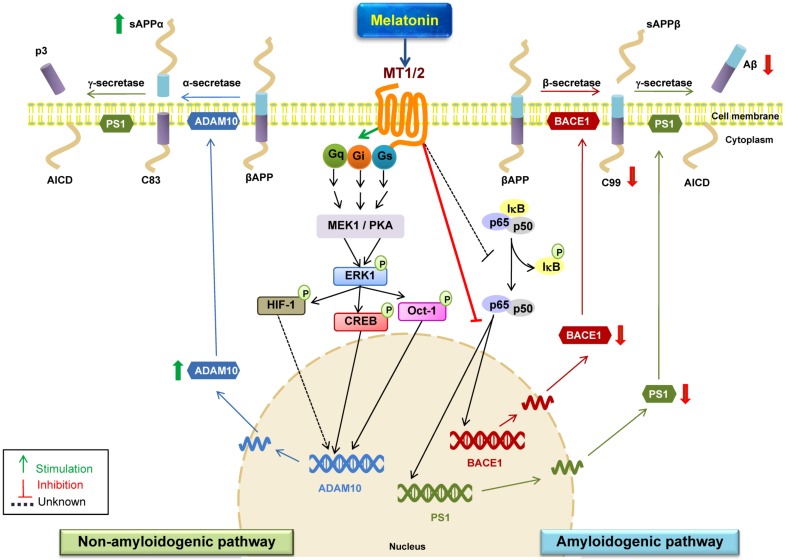
Alzheimer's disease (AD) is a chronic, progressive and prevalent neurodegenerative disease characterized by the loss of higher cognitive functions and an associated loss of memory. The thus far "incurable" stigma for AD prevails because of variations in the success rates of different treatment protocols in animal and human studies. Among the classical hypotheses explaining AD pathogenesis, the amyloid hypothesis is currently being targeted for drug development. The underlying concept is to prevent the formation of these neurotoxic peptides which play a central role in AD pathology and trigger a multispectral cascade of neurodegenerative processes post-aggregation. This could possibly be achieved by pharmacological inhibition of β- or γ-secretase or stimulating the nonamyloidogenic α-secretase. Melatonin the pineal hormone is a multifunctioning indoleamine. Production of this amphiphilic molecule diminishes with advancing age and this decrease runs parallel with the progression of AD which itself explains the potential benefits of melatonin in line of development and devastating consequences of the disease progression. Our recent studies have revealed a novel mechanism by which melatonin stimulates the nonamyloidogenic processing and inhibits the amyloidogenic processing of β-amyloid precursor protein (βAPP) by stimulating α -secretases and consequently down regulating both β- and γ-secretases at the transcriptional level. In this review, we discuss and evaluate the neuroprotective functions of melatonin in AD pathogenesis, including its role in the classical hypotheses in cellular and animal models and clinical interventions in AD patients, and suggest that with early detection, melatonin treatment is qualified to be an anti-AD therapy.
Keywords: Alzheimer's disease; aging; amyloid-β peptide; melatonin; neuroprotection.; secretases.
Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.org.
Figures
References
-
- Glenner G.G., Wong C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120(3):885–890. [http://dx.doi.org/10.1016/S0006-291X(84)80190-4]. [PMID: 6375662]. - PubMed
-
- Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993;16(11):460–465. [http:// dx.doi.org/10.1016/0166-2236(93)90078-Z]. [PMID: 7507619]. - PubMed
-
- Bartus R.T., Dean R.L., III, Beer B., Lippa A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. [http://dx.doi.org/10.1126/science.7046051]. [PMID: 7046051]. - PubMed
-
- Bartus R.T. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 2000;163(2):495–529. [http://dx.doi.org/10.1006/exnr.2000.7397]. [PMID: 10833325]. - PubMed
-
- Hardeland R. Melatonin: signaling mechanisms of a pleiotropic agent. Biofactors. 2009;35(2):183–192. [http://dx.doi.org/10.1002/ biof.23]. [PMID: 19449447]. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical