

A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population
- PMID: 28295036
- PMCID: PMC5437897
- DOI: 10.1038/ejhg.2017.16
A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population
Abstract
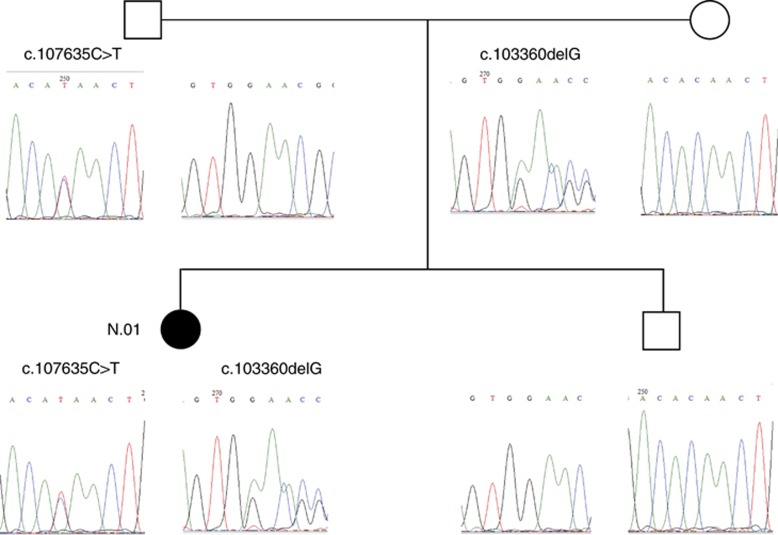
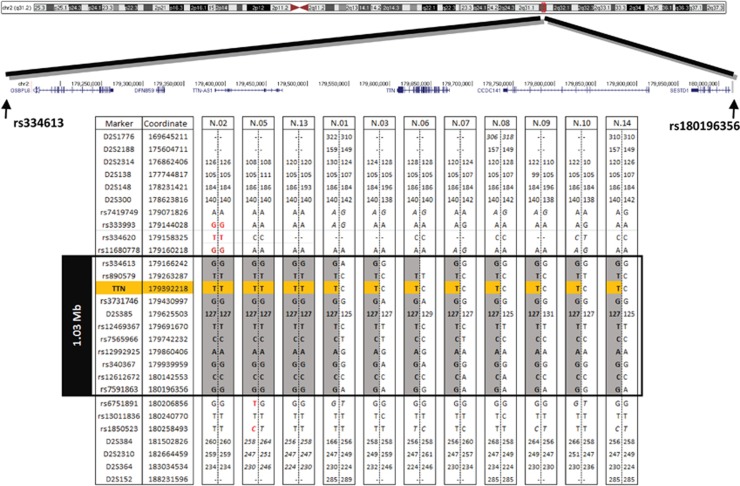
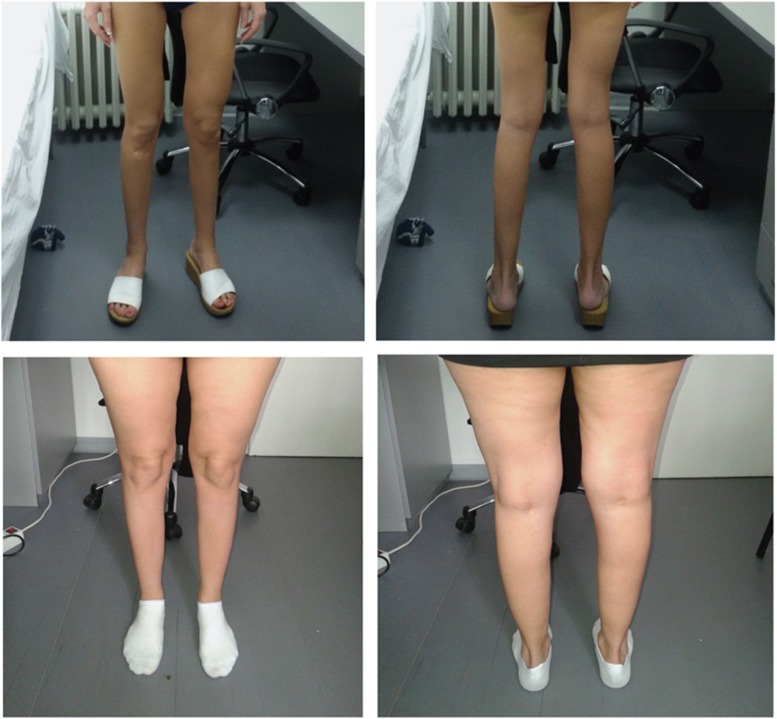
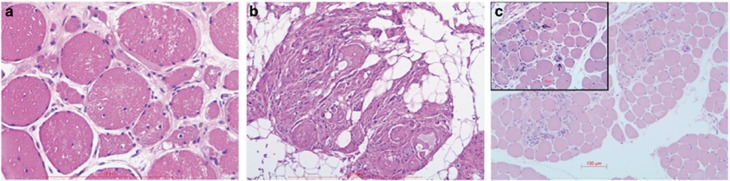
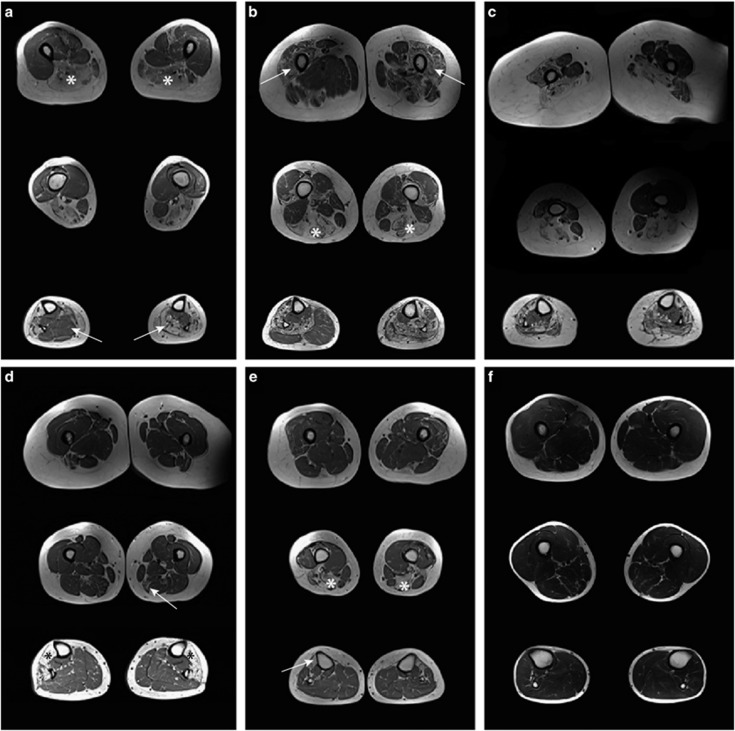
Variants in the TTN gene have been associated with distal myopathies and other distinctive phenotypes involving skeletal and cardiac muscle. Through whole-exome sequencing we identified a novel stop-gain variant (c.107635C>T, p.(Gln35879Ter)) in the TTN gene, coding a part of the M-line of titin, in 14 patients with autosomal recessive distal myopathy and Serbian ancestry. All patients share a common 1 Mb core haplotype associated with c.107635C>T, suggesting a founder variant. In compound heterozygotes, nine other TTN variants were identified: four stop-gain, three frameshift, one missense and one splice donor variant. Patients homozygous for the common variant did not show significant clinical differences to the compound heterozygous patients. The clinical presentation of all patients was an adult onset distal myopathy with predominant lower limb involvement. In addition, most patients had normal to mildly elevated serum creatine kinase levels, myopathic electromyograms, normal cardiologic and respiratory tests and muscle pathology consistent with a dystrophic process. In this study, we describe a distinct phenotype for patients with distal myopathy associated with novel recessive TTN variants including a Serbian founder variant. Our results expand the phenotypic and genetic spectrum of titinopathies and will facilitate the diagnosis of this condition in patients of Serbian origin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Udd B Dystal myopathies; in Karpati G, Hiltin-Johnes D, Bushby K, Griggs C (eds): Disorders of Voluntary Muscle. Cambridge University Press: Cambridge, UK, 2010, pp 35–40.
-
- Udd B: Molecular biology of distal mucular distrophies – sarcomeric proteins on top. Biochim Biophys Acta 2007; 1772: 145–158. - PubMed
-
- Chauveau C, Rowell J, Ferreiro A: A rising titan: TTN review and mutation update. Hum Mutat 2014; 35: 1046–1059. - PubMed
-
- Lek M, MacArthur DG: The challenge of next generation sequencing in the context of neuromuscular diseases. J Neuromuscul Dis 2014; 1: 135–149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
