

De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies
- PMID: 28296084
- PMCID: PMC7033032
- DOI: 10.1002/ajmg.a.38207
De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies
Abstract
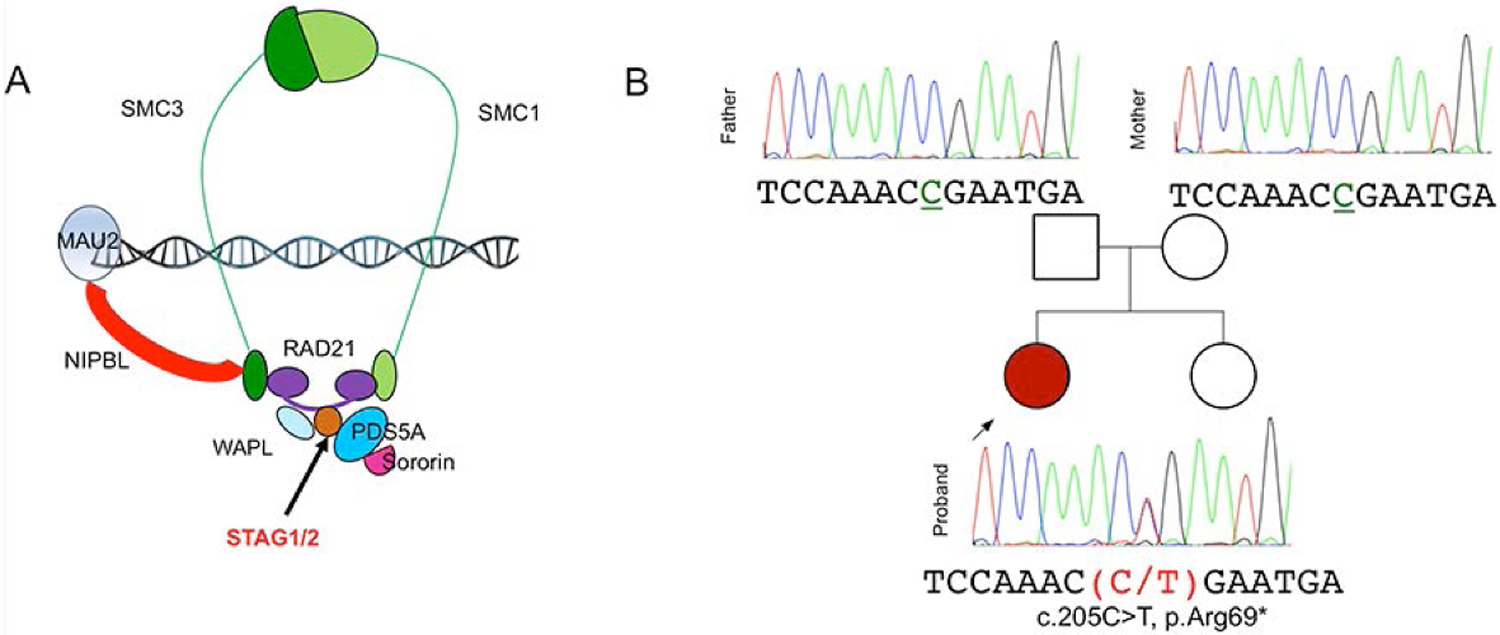
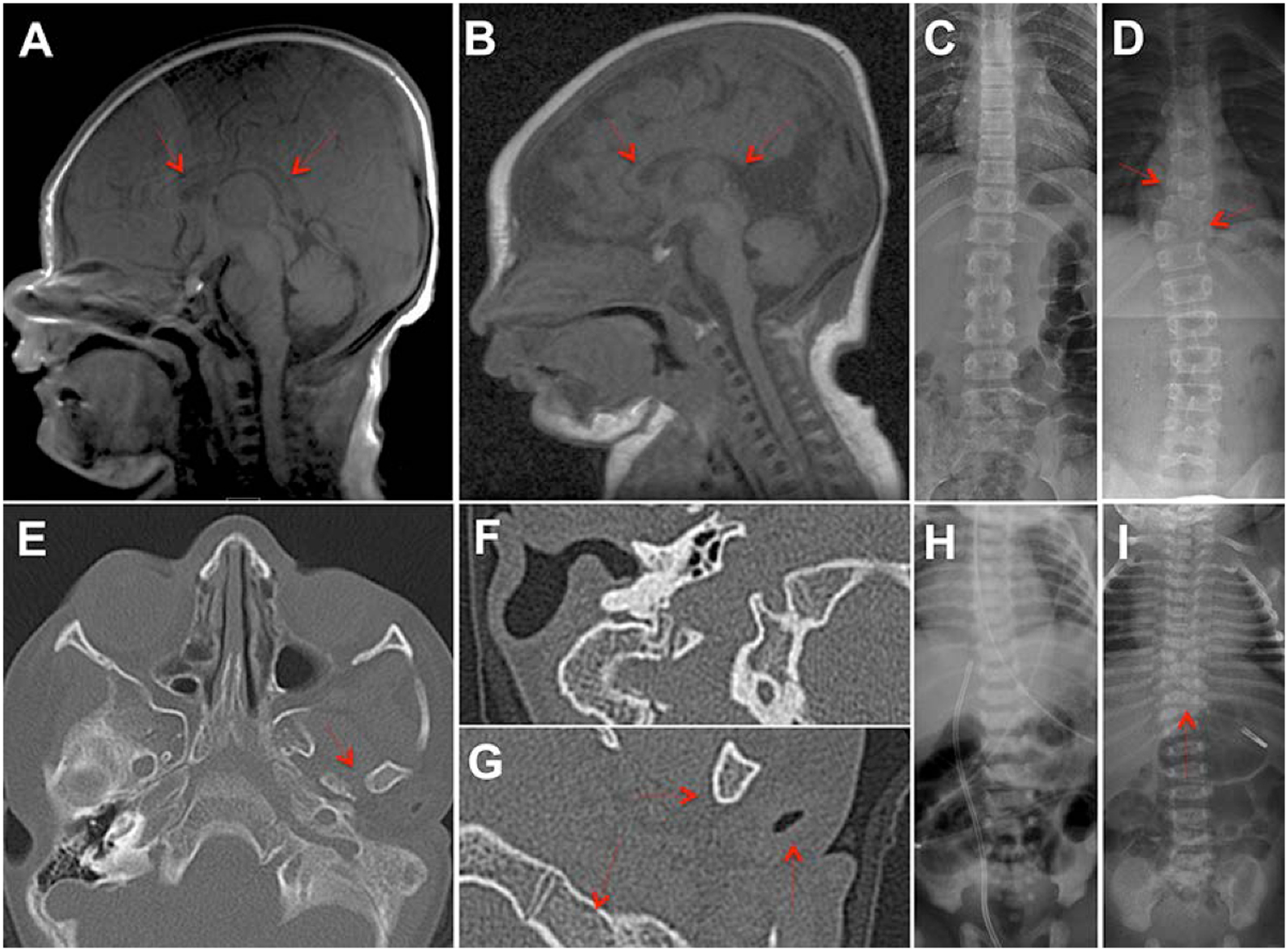
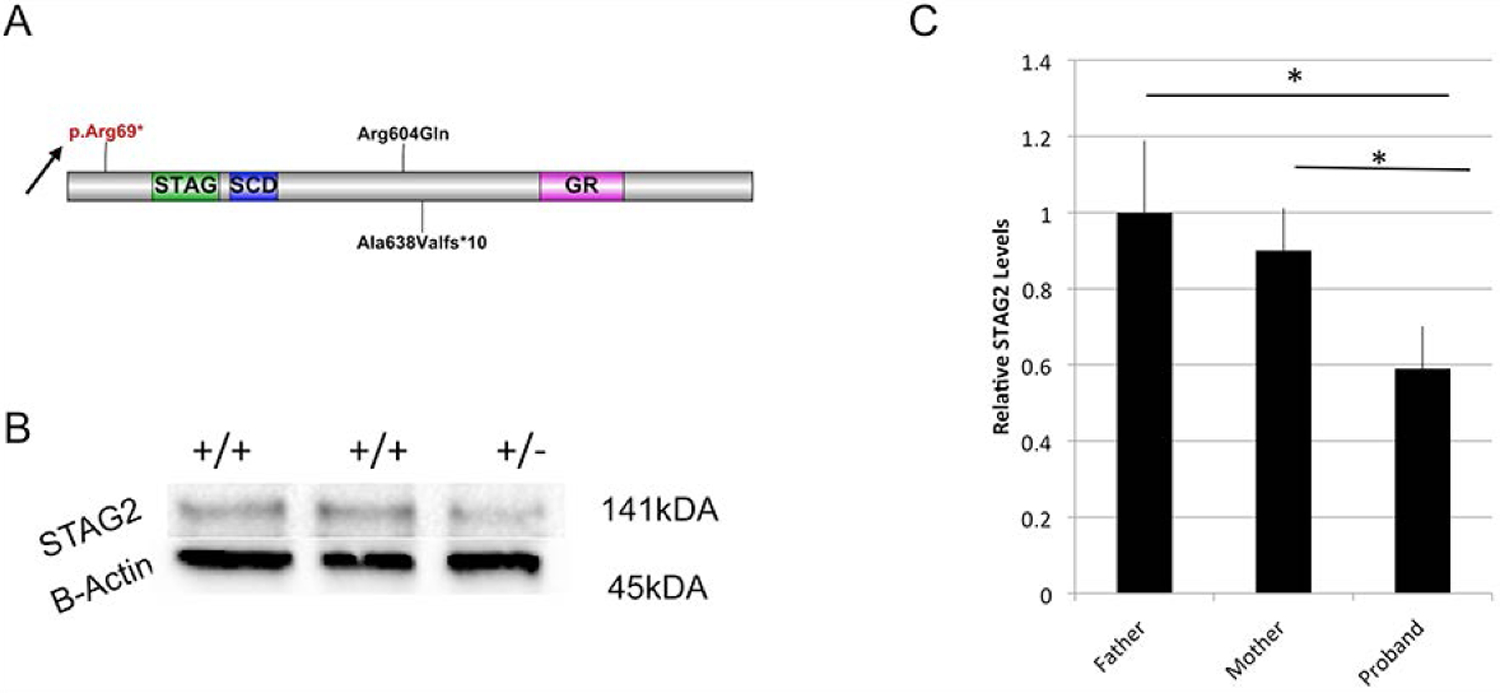
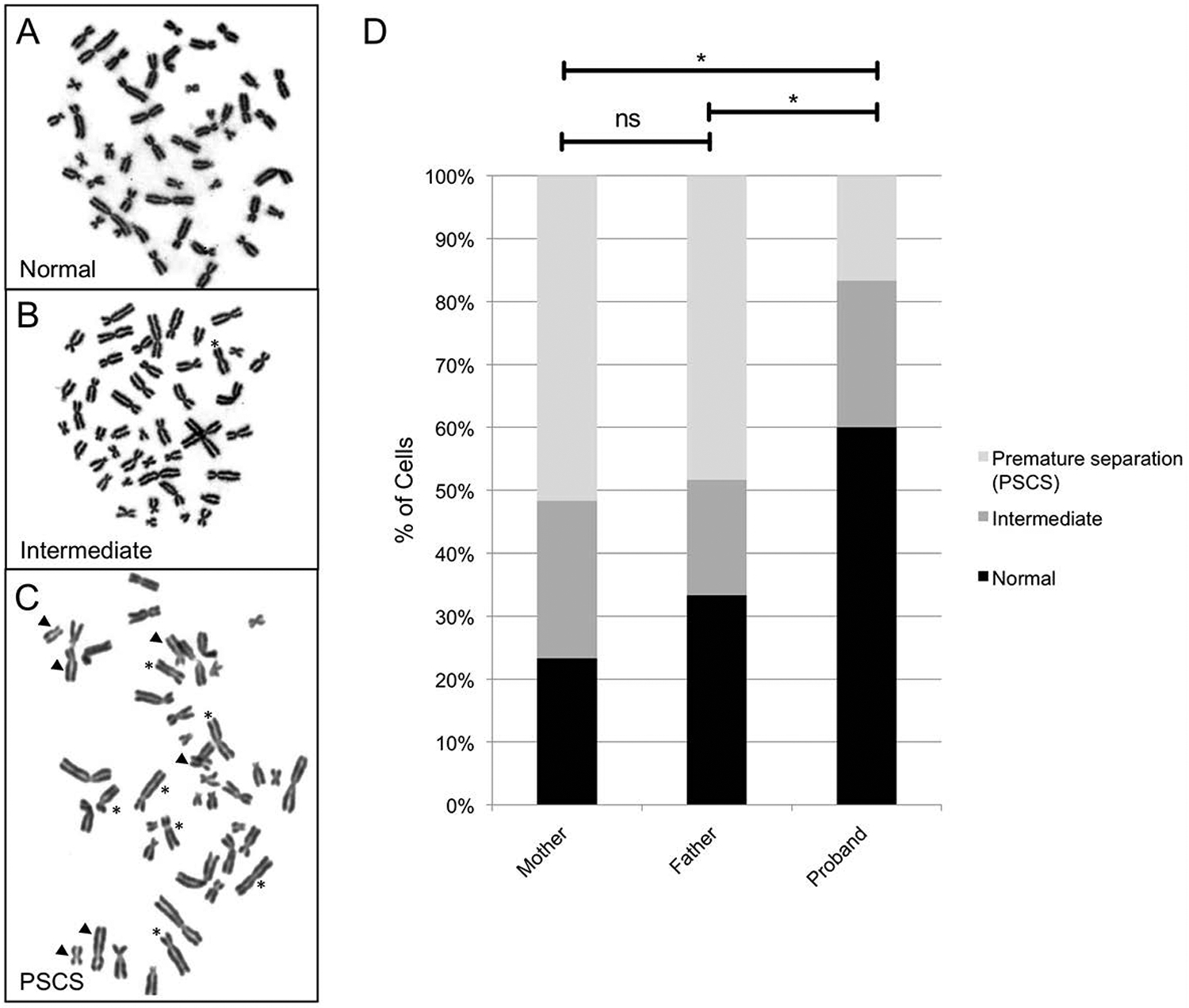
The cohesin complex is an evolutionarily conserved multi-subunit protein complex which regulates sister chromatid cohesion during mitosis and meiosis. Additionally, the cohesin complex regulates DNA replication, DNA repair, and transcription. The core of the complex consists of four subunits: SMC1A, SMC3, RAD21, and STAG1/2. Loss-of-function mutations in many of these proteins have been implicated in human developmental disorders collectively termed "cohesinopathies." Through clinical exome sequencing (CES) of an 8-year-old girl with a clinical history of global developmental delay, microcephaly, microtia with hearing loss, language delay, ADHD, and dysmorphic features, we describe a heterozygous de novo variant (c.205C>T; p.(Arg69*)) in the integral cohesin structural protein, STAG2. This variant is associated with decreased STAG2 protein expression. The analyses of metaphase spreads did not exhibit premature sister chromatid separation; however, delayed sister chromatid cohesion was observed. To further support the pathogenicity of STAG2 variants, we identified two additional female cases from the DECIPHER research database with mutations in STAG2 and phenotypes similar to our patient. Interestingly, the clinical features of these three cases are remarkably similar to those observed in other well-established cohesinopathies. Herein, we suggest that STAG2 is a dosage-sensitive gene and that heterozygous loss-of-function variants lead to a cohesinopathy.
Keywords: cohesin complex; STAG2; X-linked; cohesin-associated genes; cohesinopathy; gene dosage.
© 2017 Wiley Periodicals, Inc.
Conflict of interest statement
Figures
References
-
- Bonnet C, Leheup B, Beri M, Philippe C, Gregoire MJ, Jonveaux P. 2009. Aberrant GRIA3 transcripts with multi-exon duplications in a family with X-linked mental retardation. Am J Med Genet A 149A(6):1280–1289. - PubMed
-
- Chae JH, Hwang H, Hwang YS, Cheong HJ, Kim KJ. 2004. Influence of MECP2 gene mutation and X-chromosome inactivation on the Rett syndrome phenotype. J Child Neurol 19(7):503–508. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
