

Root microbiota drive direct integration of phosphate stress and immunity
- PMID: 28297714
- PMCID: PMC5364063
- DOI: 10.1038/nature21417
Root microbiota drive direct integration of phosphate stress and immunity
Abstract
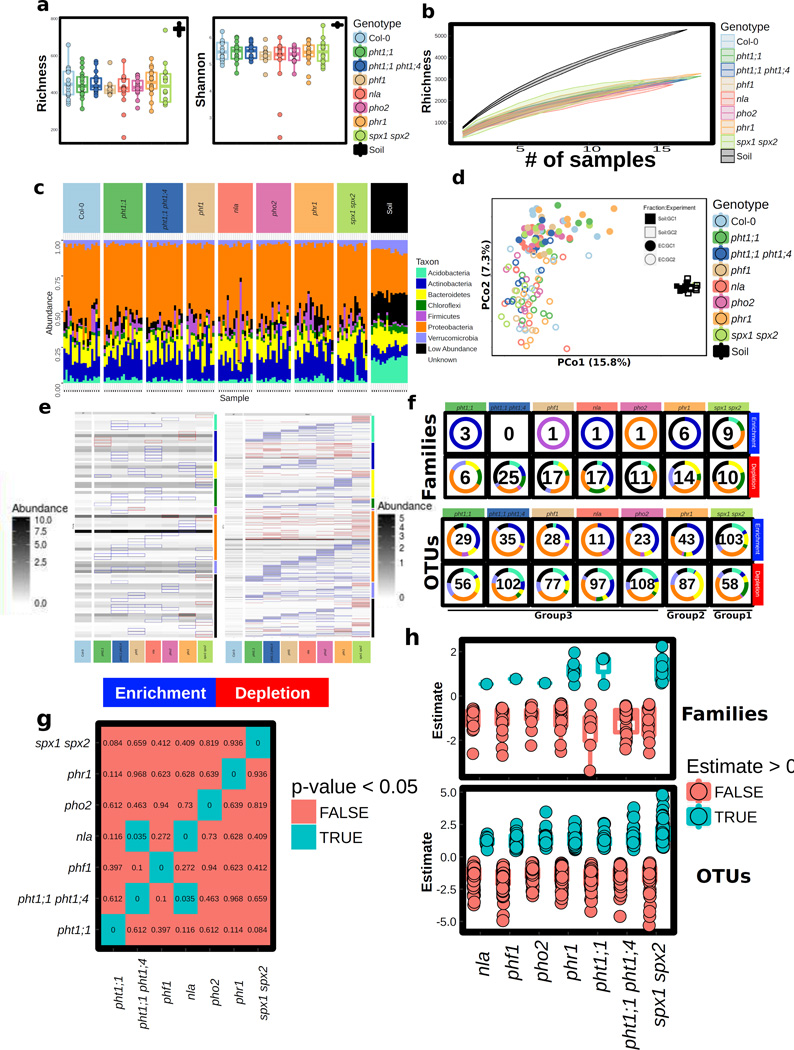
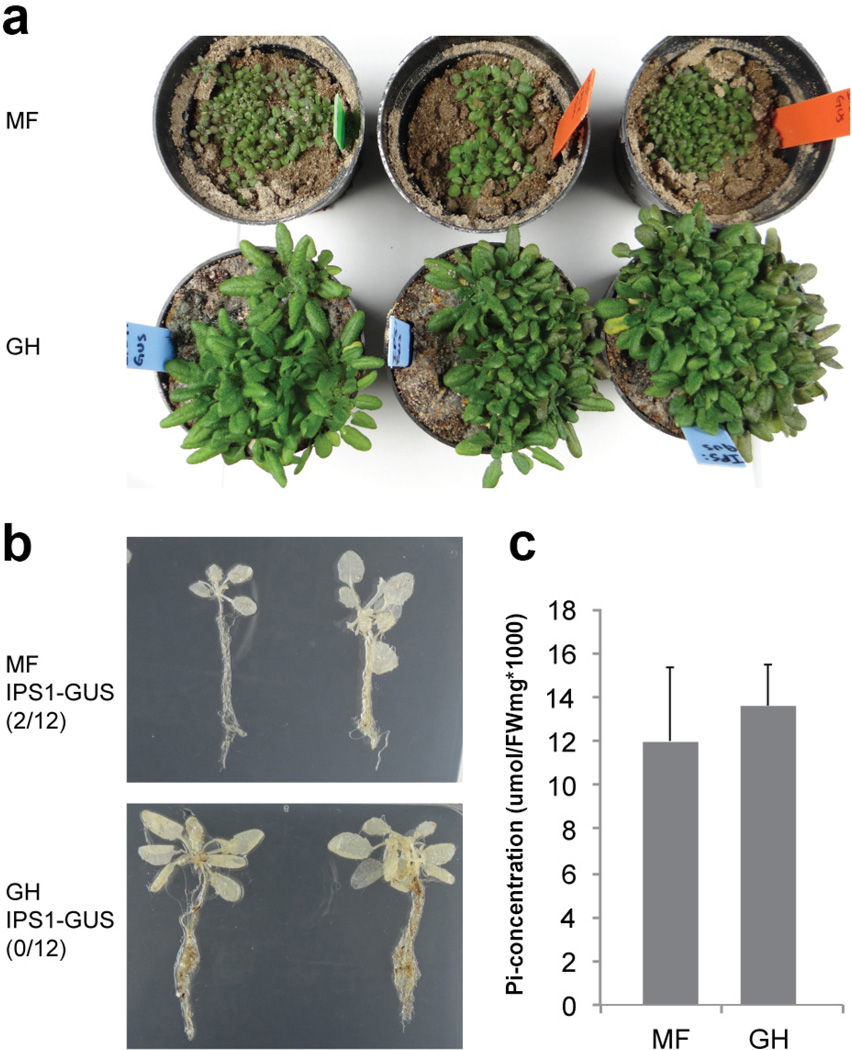
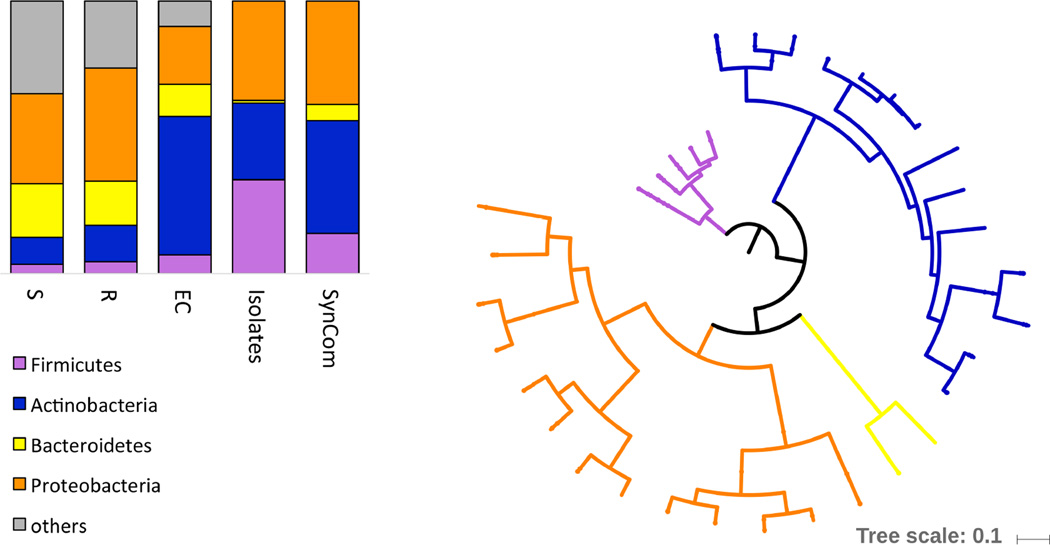
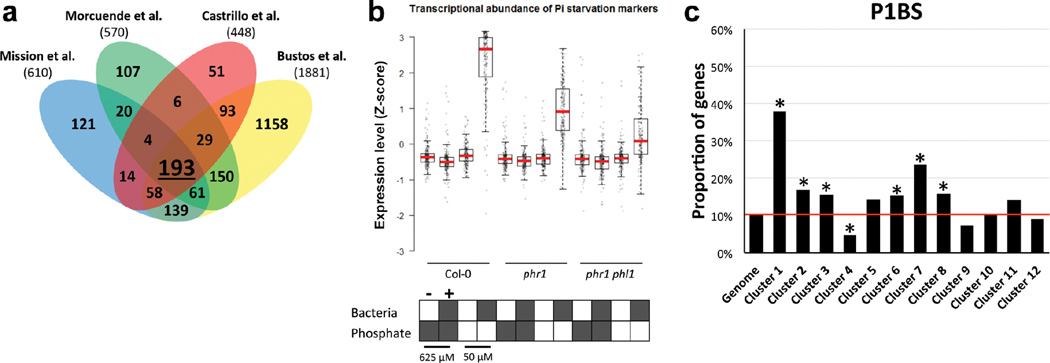
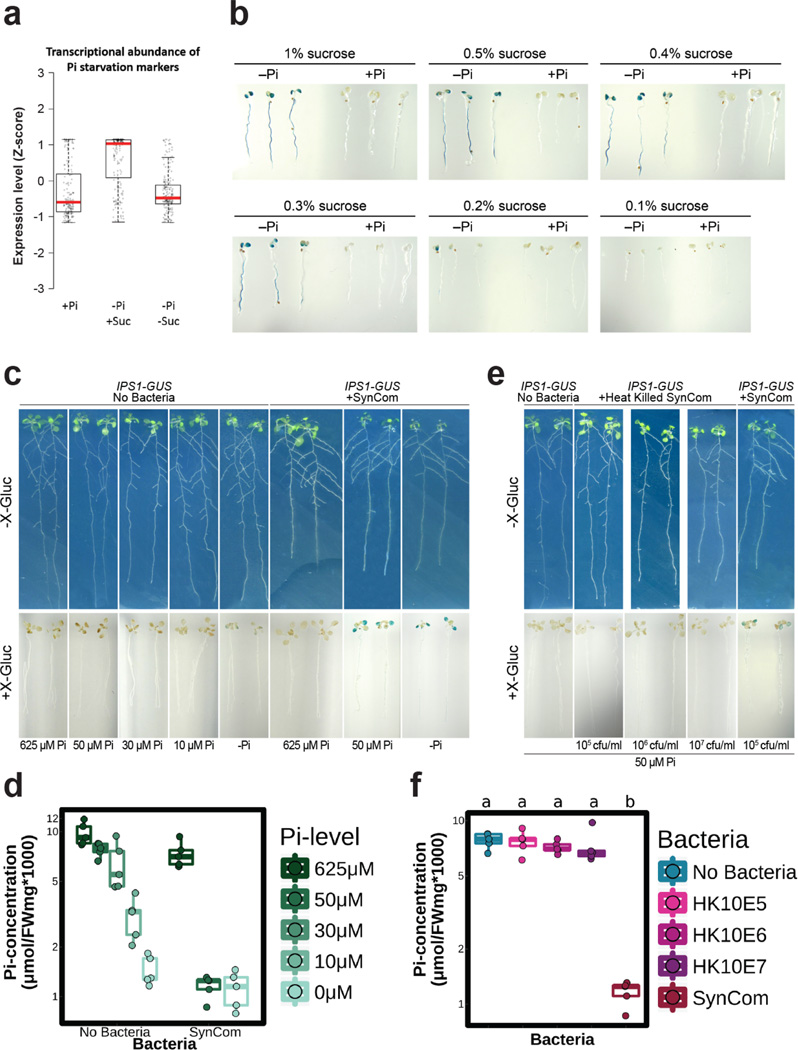
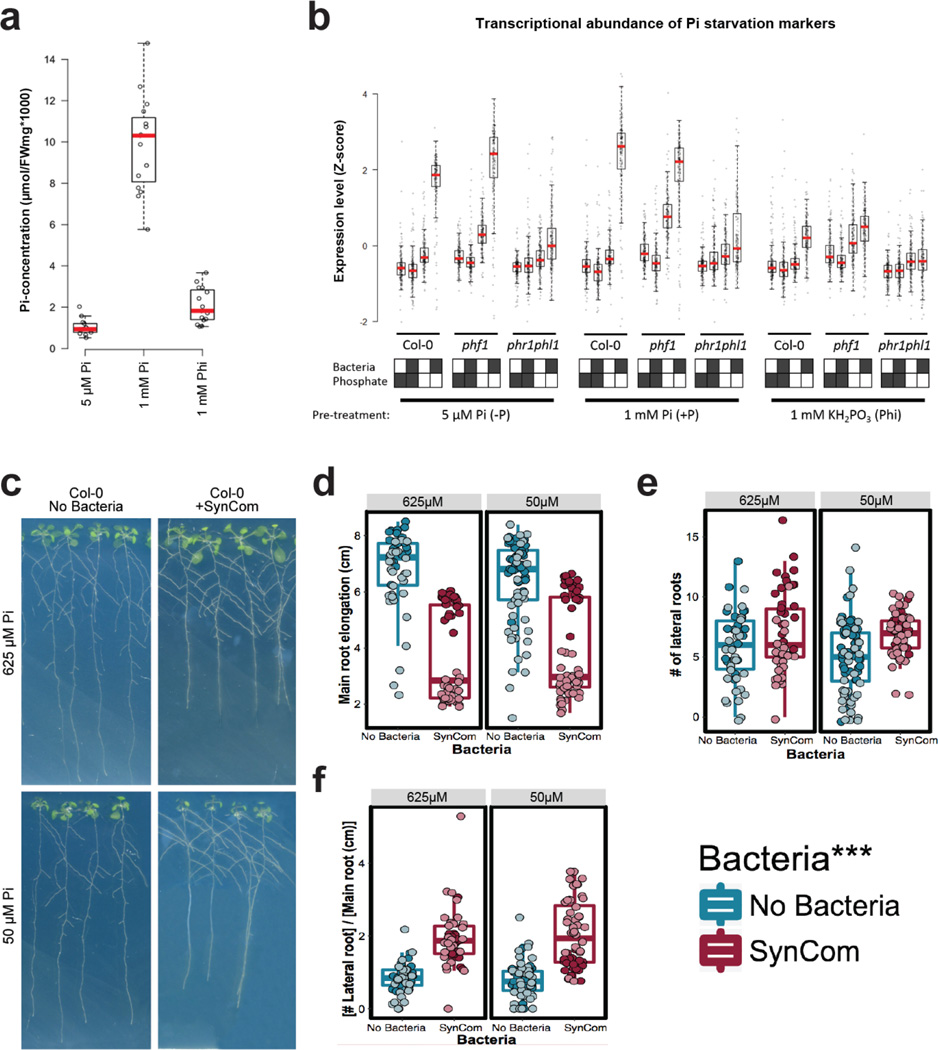
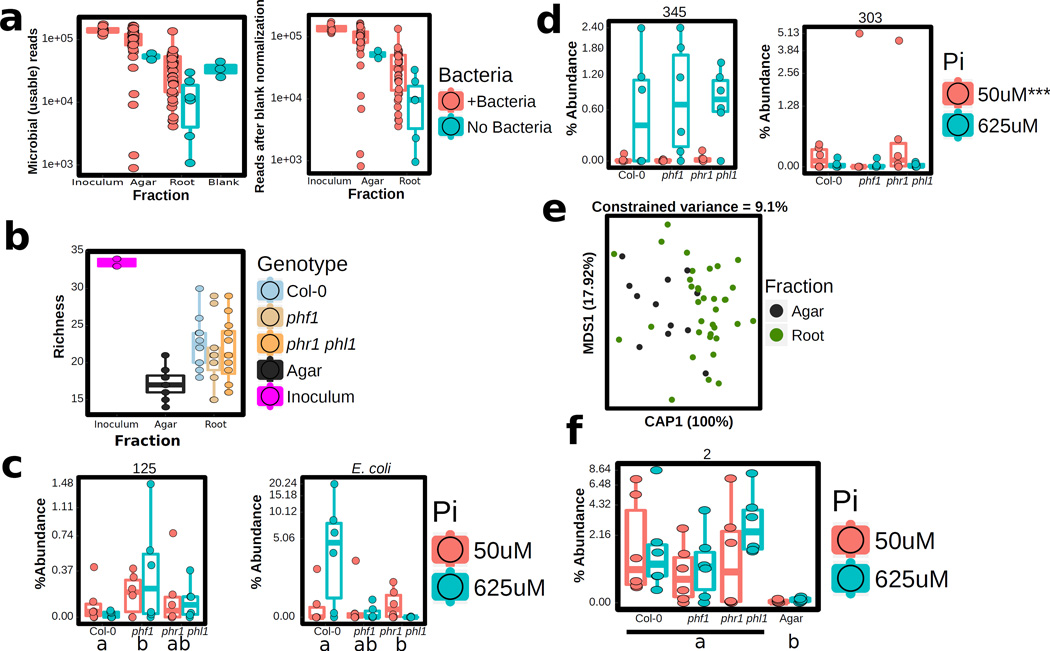
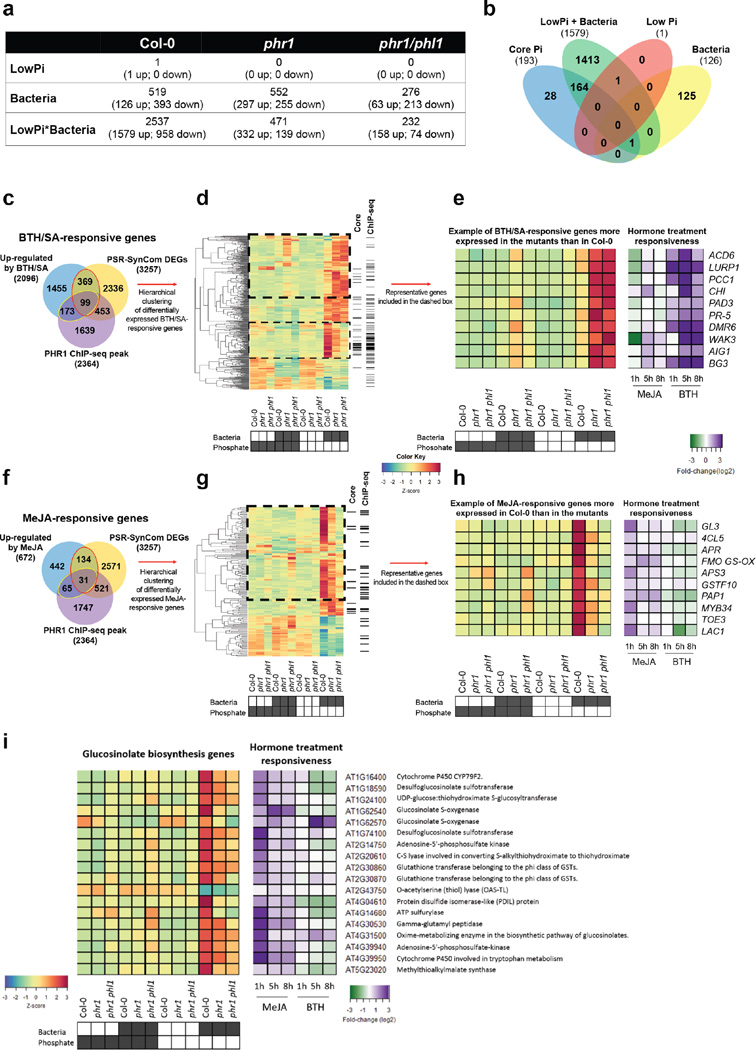
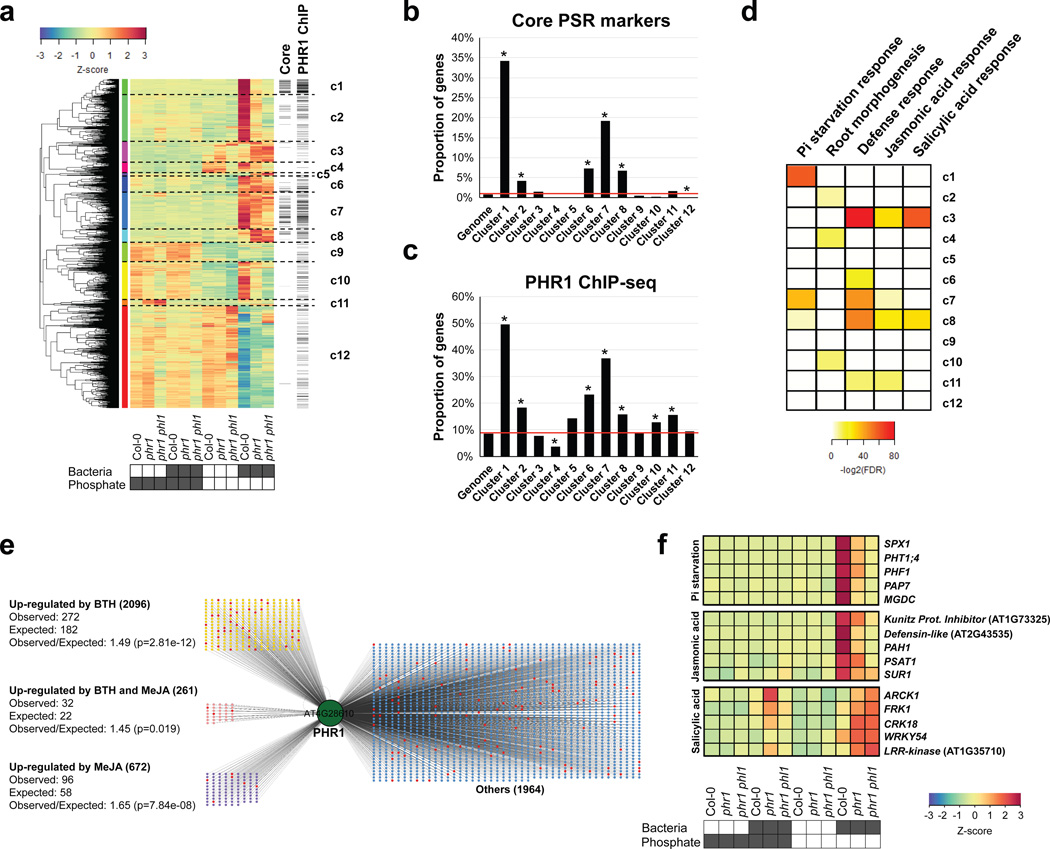
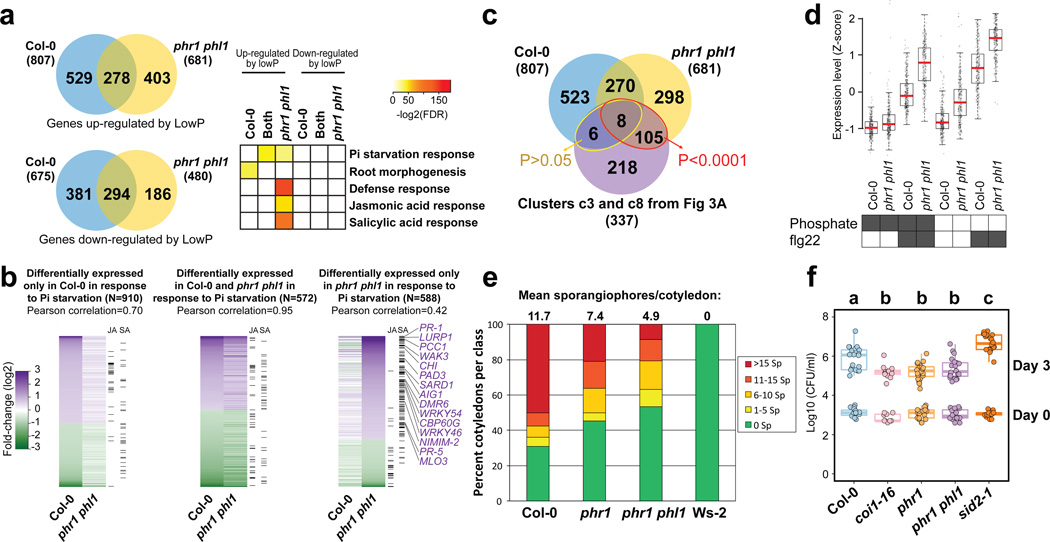
Plants live in biogeochemically diverse soils with diverse microbiota. Plant organs associate intimately with a subset of these microbes, and the structure of the microbial community can be altered by soil nutrient content. Plant-associated microbes can compete with the plant and with each other for nutrients, but may also carry traits that increase the productivity of the plant. It is unknown how the plant immune system coordinates microbial recognition with nutritional cues during microbiome assembly. Here we establish that a genetic network controlling the phosphate stress response influences the structure of the root microbiome community, even under non-stress phosphate conditions. We define a molecular mechanism regulating coordination between nutrition and defence in the presence of a synthetic bacterial community. We further demonstrate that the master transcriptional regulators of phosphate stress response in Arabidopsis thaliana also directly repress defence, consistent with plant prioritization of nutritional stress over defence. Our work will further efforts to define and deploy useful microbes to enhance plant performance.
Figures
Comment in
-
PHR1 Balances between Nutrition and Immunity in Plants.Dev Cell. 2017 Apr 10;41(1):5-7. doi: 10.1016/j.devcel.2017.03.019. Dev Cell. 2017. PMID: 28399400
References
-
- Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013;64:807–838. - PubMed
-
- Lebeis SL, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–864. - PubMed
-
- Hacquard S, et al. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe. 2015;17:603–616. - PubMed
-
- Zhu Q, Riley WJ, Tang J, Koven CD. Multiple soil nutrient competition between plants, microbes, and mineral surfaces: model development, parameterization, and example applications in several tropical forests. Biogeosciences. 2016;13:341–363.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
