

CLImAT-HET: detecting subclonal copy number alterations and loss of heterozygosity in heterogeneous tumor samples from whole-genome sequencing data
- PMID: 28298214
- PMCID: PMC5351278
- DOI: 10.1186/s12920-017-0255-4
CLImAT-HET: detecting subclonal copy number alterations and loss of heterozygosity in heterogeneous tumor samples from whole-genome sequencing data
Abstract
Background: Copy number alterations (CNA) and loss of heterozygosity (LOH) represent a large proportion of genetic structural variations of cancer genomes. These aberrations are continuously accumulated during the procedure of clonal evolution and patterned by phylogenetic branching. This invariably results in the emergence of multiple cell populations with distinct complement of mutational landscapes in tumor sample. With the advent of next-generation sequencing technology, inference of subclonal populations has become one of the focused interests in cancer-associated studies, and is usually based on the assessment of combinations of somatic single-nucleotide variations (SNV), CNA and LOH. However, cancer samples often have several inherent issues, such as contamination of normal stroma, tumor aneuploidy and intra-tumor heterogeneity. Addressing these critical issues is imperative for accurate profiling of clonal architecture.
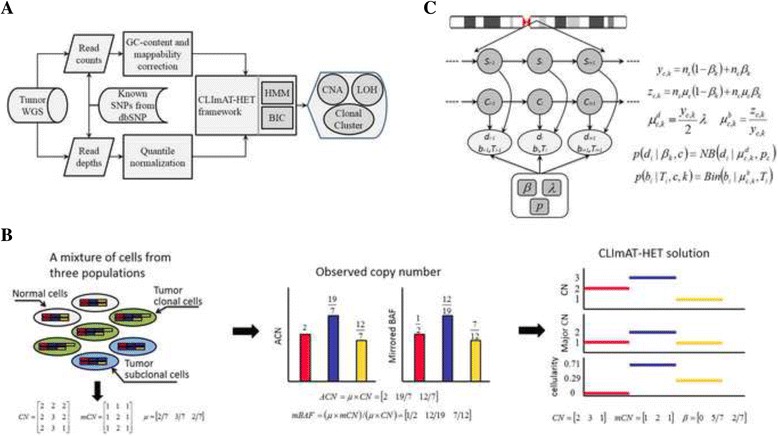
Methods: We present CLImAT-HET, a computational method designed for capturing clonal diversity in the CNA/LOH dimensions by taking into account the intra-tumor heterogeneity issue, in the case where a reference or matched normal sample is absent. The algorithm quantitatively represents the clonal identification problem using a factorial hidden Markov model, and takes an integrated analysis of read counts and allele frequency data. It is able to infer subclonal CNA and LOH events as well as the fraction of cells harboring each event.
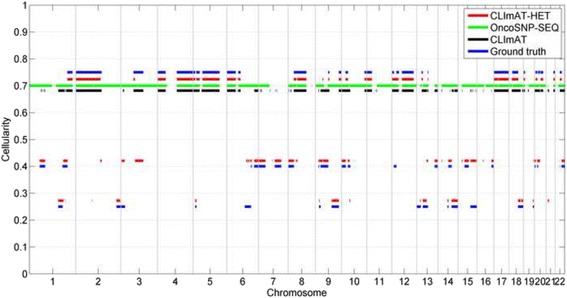
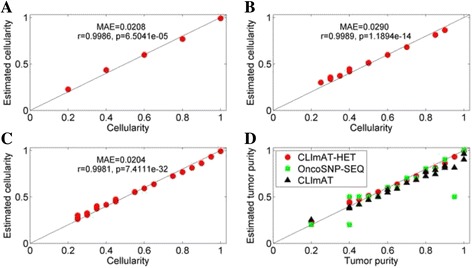
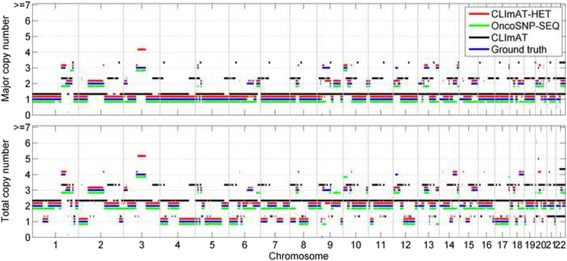
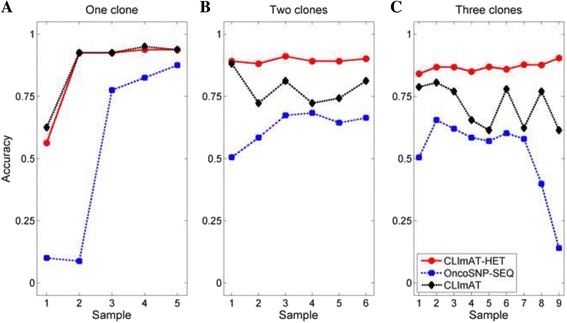
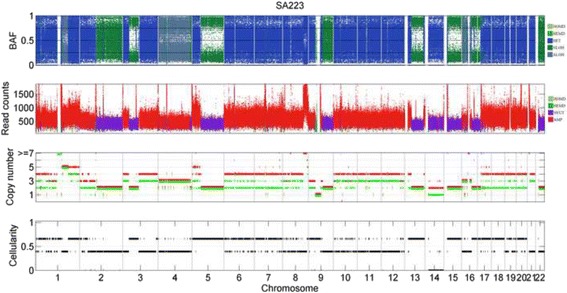
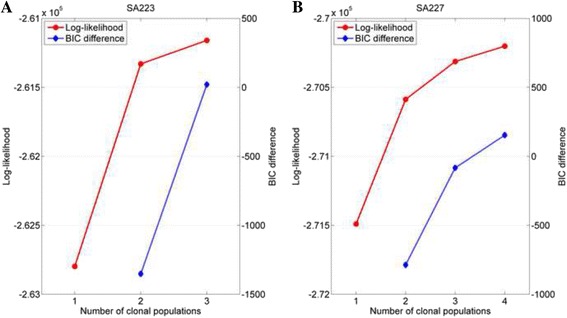
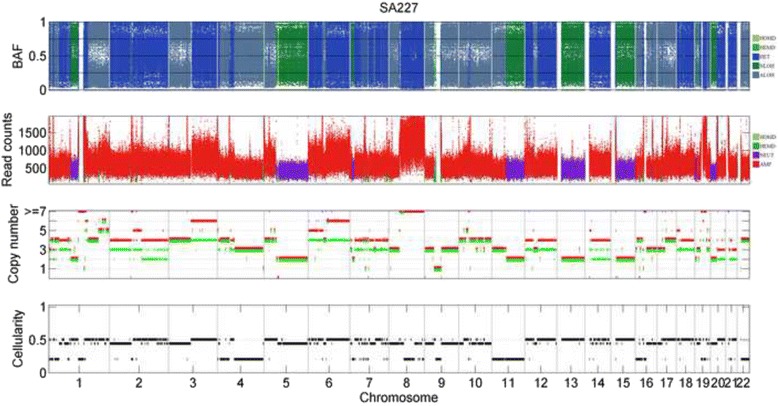
Results: The results on simulated datasets indicate that CLImAT-HET has high power to identify CNA/LOH segments, it achieves an average accuracy of 0.87. It can also accurately infer proportion of each clonal population with an overall Pearson correlation coefficient of 0.99 and a mean absolute error of 0.02. CLImAT-HET shows significant advantages when compared with other existing methods. Application of CLImAT-HET to 5 primary triple negative breast cancer samples demonstrates its ability to capture clonal diversity in the CAN/LOH dimensions. It detects two clonal populations in one sample, and three clonal populations in one other sample.
Conclusions: CLImAT-HET, a novel algorithm is introduced to infer CNA/LOH segments from heterogeneous tumor samples. We demonstrate CLImAT-HET's ability to accurately recover clonal compositions using tumor WGS data without a match normal sample.
Keywords: Bayesian information criterion; Copy number alteration; Hidden Markov model; Intra-tumor heterogeneity; Loss of heterozygosity; Whole-genome sequencing.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
