

Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System
- PMID: 28298428
- PMCID: PMC5539405
- DOI: 10.1152/physrev.00010.2016
Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System
Abstract
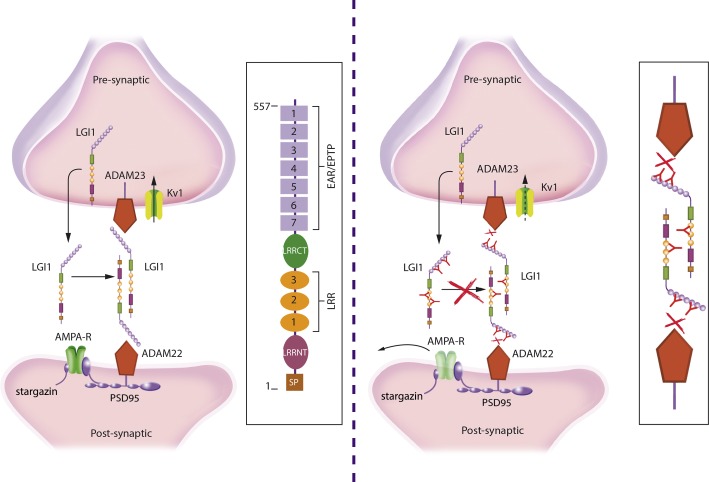
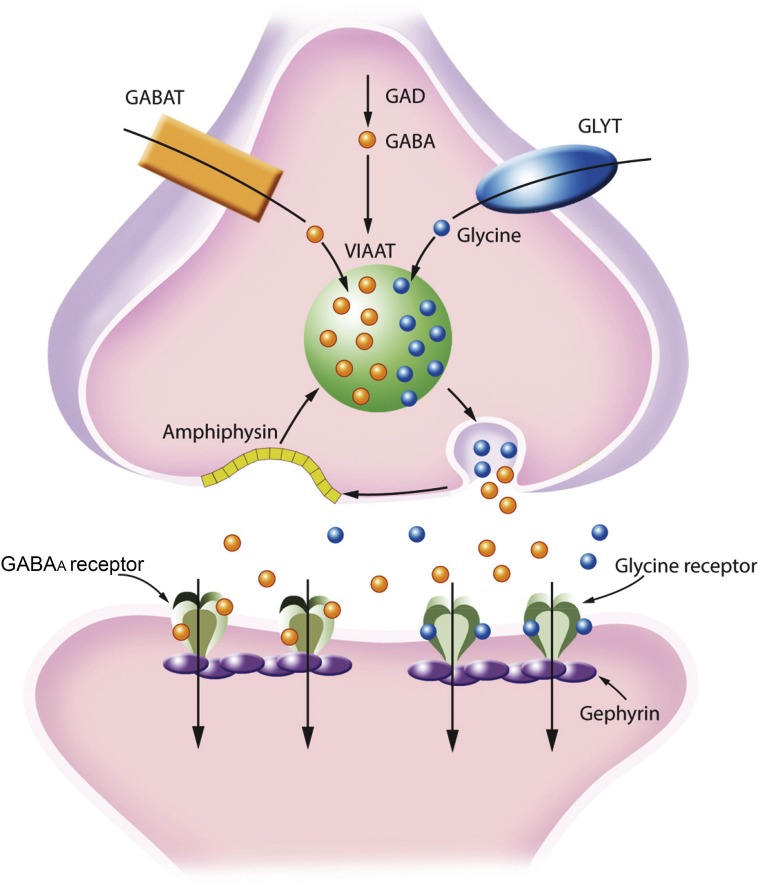
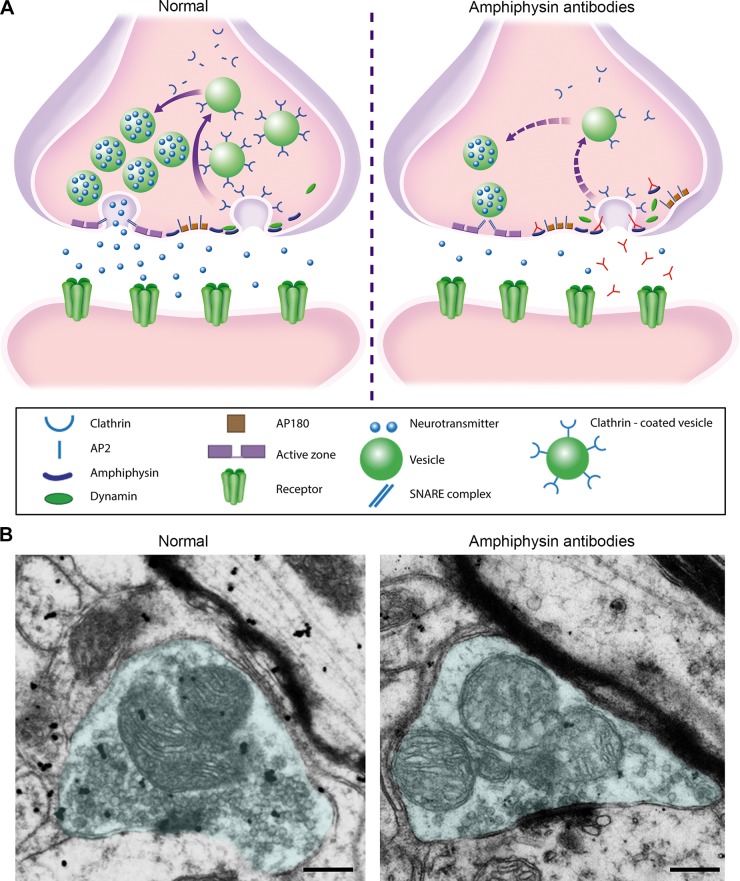
Investigations in the last 10 years have revealed a new category of neurological diseases mediated by antibodies against cell surface and synaptic proteins. There are currently 16 such diseases all characterized by autoantibodies against neuronal proteins involved in synaptic signaling and plasticity. In clinical practice these findings have changed the diagnostic and treatment approach to potentially lethal, but now treatable, neurological and psychiatric syndromes previously considered idiopathic or not even suspected to be immune-mediated. Studies show that patients' antibodies can impair the surface dynamics of the target receptors eliminating them from synapses (e.g., NMDA receptor), block the function of the antigens without changing their synaptic density (e.g., GABAb receptor), interfere with synaptic protein-protein interactions (LGI1, Caspr2), alter synapse formation (e.g., neurexin-3α), or by unclear mechanisms associate to a new form of tauopathy (IgLON5). Here we first trace the process of discovery of these diseases, describing the triggers and symptoms related to each autoantigen, and then review in detail the structural and functional alterations caused by the autoantibodies with special emphasis in those (NMDA receptor, amphiphysin) that have been modeled in animals.
Copyright © 2017 the American Physiological Society.
Figures


















References
-
- Abou-Zeid E, Boursoulian LJ, Metzer WS, Gundogdu B. Morvan syndrome: a case report and review of the literature. J Clin Neuromuscul Dis 13: 214–227, 2012. - PubMed
-
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9: 387, 2008. - PubMed
-
- Adams C, Diadori P, Schoenroth L, Fritzler M. Autoantibodies in childhood post-varicella acute cerebellar ataxia. Can J Neurol Sci 27: 316–320, 2000. - PubMed
-
- Alamowitch S, Graus F, Uchuya M, Rene R, Bescansa E, Delattre JY. Limbic encephalitis and small cell lung cancer. Clinical and immunological features. Brain 120: 923–928, 1997. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical