

Prion Strain Characterization of a Novel Subtype of Creutzfeldt-Jakob Disease
- PMID: 28298604
- PMCID: PMC5432879
- DOI: 10.1128/JVI.02390-16
Prion Strain Characterization of a Novel Subtype of Creutzfeldt-Jakob Disease
Abstract
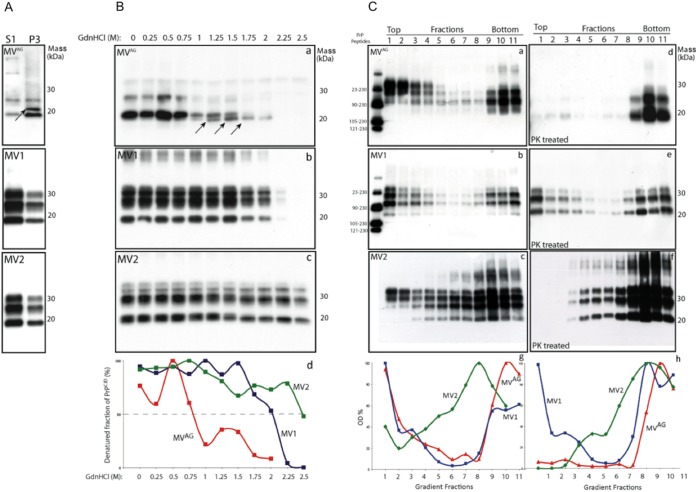
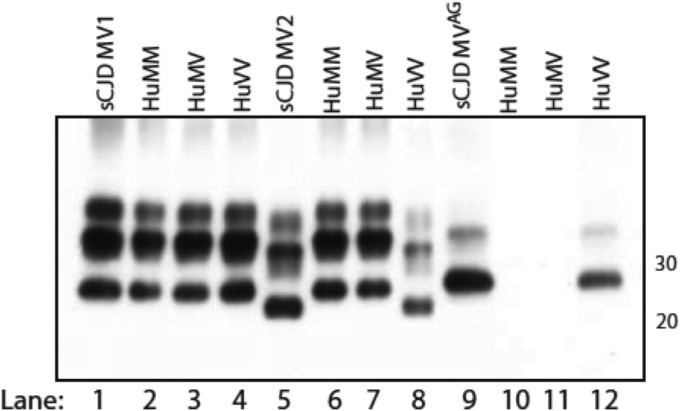
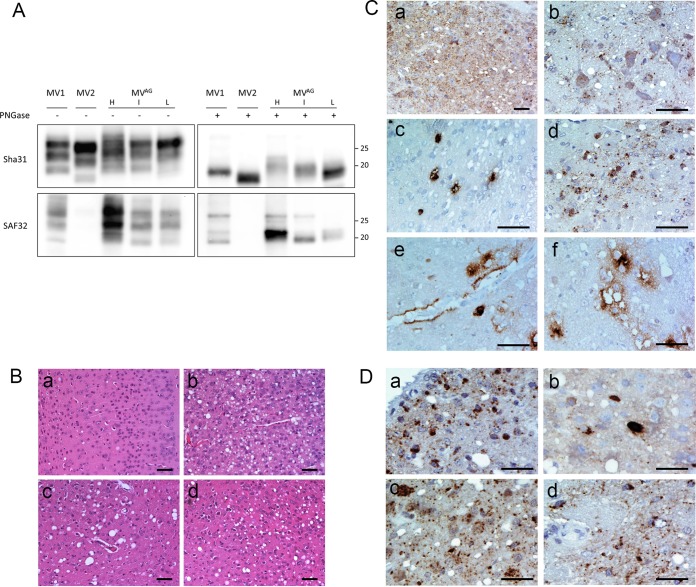
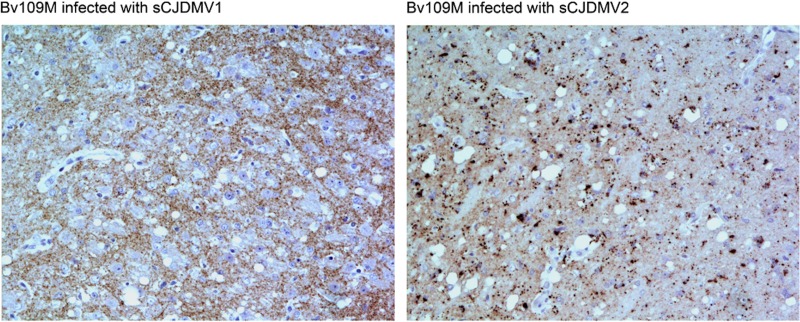
In 2007, we reported a patient with an atypical form of Creutzfeldt-Jakob disease (CJD) heterozygous for methionine-valine (MV) at codon 129 who showed a novel pathological prion protein (PrPTSE) conformation with an atypical glycoform (AG) profile and intraneuronal PrP deposition. In the present study, we further characterize the conformational properties of this pathological prion protein (PrPTSE MVAG), showing that PrPTSE MVAG is composed of multiple conformers with biochemical properties distinct from those of PrPTSE type 1 and type 2 of MV sporadic CJD (sCJD). Experimental transmission of CJD-MVAG to bank voles and gene-targeted transgenic mice carrying the human prion protein gene (TgHu mice) showed unique transmission rates, survival times, neuropathological changes, PrPTSE deposition patterns, and PrPTSE glycotypes that are distinct from those of sCJD-MV1 and sCJD-MV2. These biochemical and experimental data suggest the presence of a novel prion strain in CJD-MVAGIMPORTANCE Sporadic Creutzfeldt-Jakob disease is caused by the misfolding of the cellular prion protein, which assumes two different major conformations (type 1 and type 2) and, together with the methionine/valine polymorphic codon 129 of the prion protein gene, contribute to the occurrence of distinct clinical-pathological phenotypes. Inoculation in laboratory rodents of brain tissues from the six possible combinations of pathological prion protein types with codon 129 genotypes results in the identification of 3 or 4 strains of prions. We report on the identification of a novel strain of Creutzfeldt-Jakob disease isolated from a patient who carried an abnormally glycosylated pathological prion protein. This novel strain has unique biochemical characteristics, does not transmit to humanized transgenic mice, and shows exclusive transmission properties in bank voles. The identification of a novel human prion strain improves our understanding of the pathogenesis of the disease and of possible mechanisms of prion transmission.
Keywords: Creutzfeldt-Jakob disease; humanized mice; prion strain; prions.
Copyright © 2017 American Society for Microbiology.
Figures
Similar articles
-
Prions from Sporadic Creutzfeldt-Jakob Disease Patients Propagate as Strain Mixtures.mBio. 2020 Jun 16;11(3):e00393-20. doi: 10.1128/mBio.00393-20. mBio. 2020. PMID: 32546613 Free PMC article.
-
Sensitive protein misfolding cyclic amplification of sporadic Creutzfeldt-Jakob disease prions is strongly seed and substrate dependent.Sci Rep. 2021 Feb 18;11(1):4058. doi: 10.1038/s41598-021-83630-1. Sci Rep. 2021. PMID: 33603091 Free PMC article.
-
Sporadic Creutzfeldt-Jakob disease VM1: phenotypic and molecular characterization of a novel subtype of human prion disease.Acta Neuropathol Commun. 2022 Aug 17;10(1):114. doi: 10.1186/s40478-022-01415-7. Acta Neuropathol Commun. 2022. PMID: 35978418 Free PMC article.
-
Neuropathological and biochemical criteria to identify acquired Creutzfeldt-Jakob disease among presumed sporadic cases.Neuropathology. 2016 Jun;36(3):305-10. doi: 10.1111/neup.12270. Epub 2015 Dec 15. Neuropathology. 2016. PMID: 26669818 Review.
-
Pathological spectrum of sporadic Creutzfeldt-Jakob disease.Pathology. 2025 Mar;57(2):196-206. doi: 10.1016/j.pathol.2024.09.005. Epub 2024 Nov 13. Pathology. 2025. PMID: 39665904 Review.
Cited by
-
The Zoonotic Potential of Chronic Wasting Disease-A Review.Foods. 2023 Feb 15;12(4):824. doi: 10.3390/foods12040824. Foods. 2023. PMID: 36832899 Free PMC article. Review.
-
Recent advances in the histo-molecular pathology of human prion disease.Brain Pathol. 2019 Mar;29(2):278-300. doi: 10.1111/bpa.12695. Epub 2019 Jan 22. Brain Pathol. 2019. PMID: 30588685 Free PMC article. Review.
-
Case report: Atypical young case of MV1 Creutzfeldt-Jakob disease with unusually long survival.Front Cell Neurosci. 2025 Jan 3;18:1518542. doi: 10.3389/fncel.2024.1518542. eCollection 2024. Front Cell Neurosci. 2025. PMID: 39830037 Free PMC article.
-
Prion replication environment defines the fate of prion strain adaptation.PLoS Pathog. 2018 Jun 21;14(6):e1007093. doi: 10.1371/journal.ppat.1007093. eCollection 2018 Jun. PLoS Pathog. 2018. PMID: 29928047 Free PMC article.
-
Gerstmann-Sträussler-Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles.Biomolecules. 2022 Oct 21;12(10):1537. doi: 10.3390/biom12101537. Biomolecules. 2022. PMID: 36291746 Free PMC article.
References
-
- Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, Collins S, Lewis V, Sutcliffe T, Guilivi A, Delasnerie-Laupretre N, Brandel JP, Alperovitch A, Zerr I, Poser S, Kretzschmar HA, Ladogana A, Rietvald I, Mitrova E, Martinez-Martin P, de Pedro-Cuesta J, Glatzel M, Aguzzi A, Cooper S, Mackenzie J, van Duijn CM, Will RG. 2004. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 127:2348–2359. doi:10.1093/brain/awh249. - DOI - PubMed
-
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. 1999. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
