

Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes
- PMID: 28298952
- PMCID: PMC5337354
- DOI: 10.1155/2017/6437467
Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes
Abstract
Diabetes mellitus (DM) displays a high morbidity. The diabetic heart is susceptible to myocardial ischemia/reperfusion (MI/R) injury. Impaired activation of prosurvival pathways, endoplasmic reticulum (ER) stress, increased basal oxidative state, and decreased antioxidant defense and autophagy may render diabetic hearts more vulnerable to MI/R injury. Oxidative stress and mTOR signaling crucially regulate cardiometabolism, affecting MI/R injury under diabetes. Producing reactive oxygen species (ROS) and reactive nitrogen species (RNS), uncoupling nitric oxide synthase (NOS), and disturbing the mitochondrial quality control may be three major mechanisms of oxidative stress. mTOR signaling presents both cardioprotective and cardiotoxic effects on the diabetic heart, which interplays with oxidative stress directly or indirectly. Antihyperglycemic agent metformin and newly found free radicals scavengers, Sirt1 and CTRP9, may serve as promising pharmacological therapeutic targets. In this review, we will focus on the role of oxidative stress and mTOR signaling in the pathophysiology of MI/R injury in diabetes and discuss potential mechanisms and their interactions in an effort to provide some evidence for cardiometabolic targeted therapies for ischemic heart disease (IHD).
Conflict of interest statement
The authors declare that there is no conflict of interests regarding the publication of this paper.
Figures
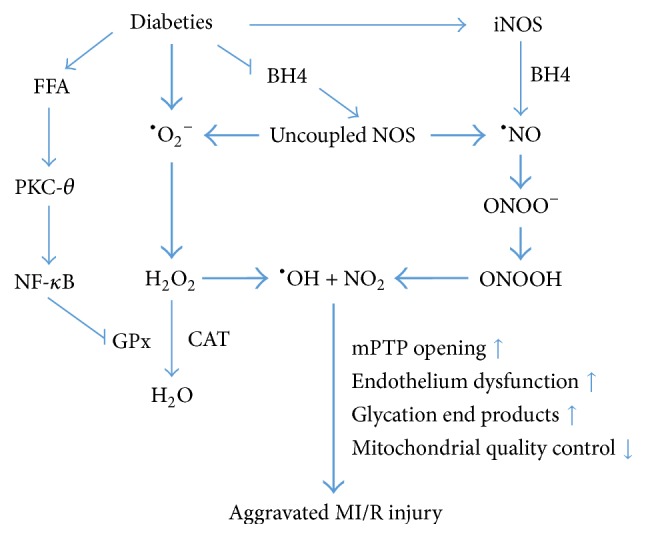
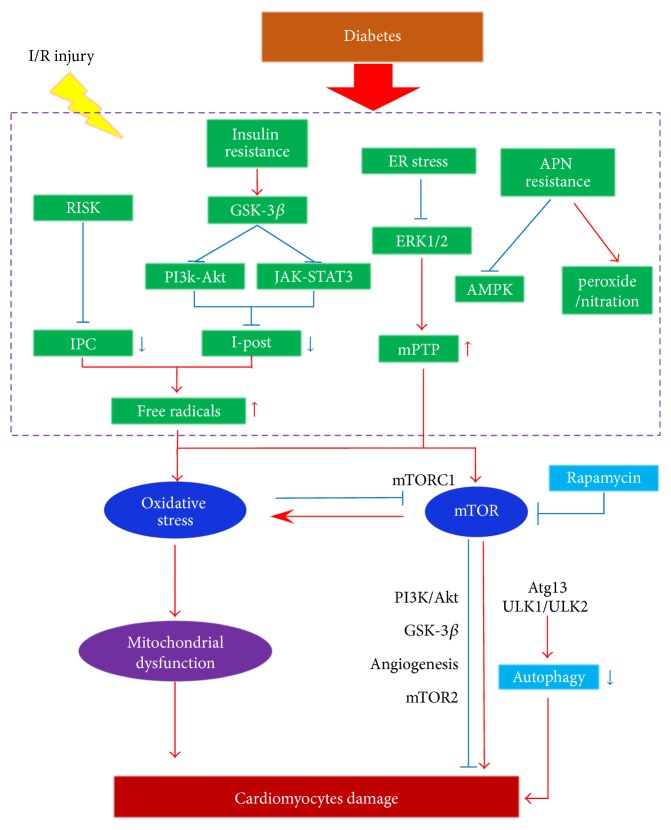
References
-
- Zoroufian A., Razmi T., Taghavi-Shavazi M., Lotfi-Tokaldany M., Jalali A. Evaluation of subclinical left ventricular dysfunction in diabetic patients: longitudinal strain velocities and left ventricular dyssynchrony by two-dimensional speckle tracking echocardiography study. Echocardiography. 2014;31(4):456–463. doi: 10.1111/echo.12389. - DOI - PubMed
-
- Shah A. M., Shin S. H., Takeuchi M., et al. Left ventricular systolic and diastolic function, remodelling, and clinical outcomes among patients with diabetes following myocardial infarction and the influence of direct renin inhibition with aliskiren. European Journal of Heart Failure. 2012;14(2):185–192. doi: 10.1093/eurjhf/hfr125. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
