

Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO)
- PMID: 28301468
- PMCID: PMC5354242
- DOI: 10.1371/journal.pone.0169687
Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO)
Erratum in
-
Correction: Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO).PLoS One. 2017 Jul 7;12(7):e0181222. doi: 10.1371/journal.pone.0181222. eCollection 2017. PLoS One. 2017. PMID: 28686717 Free PMC article.
Abstract
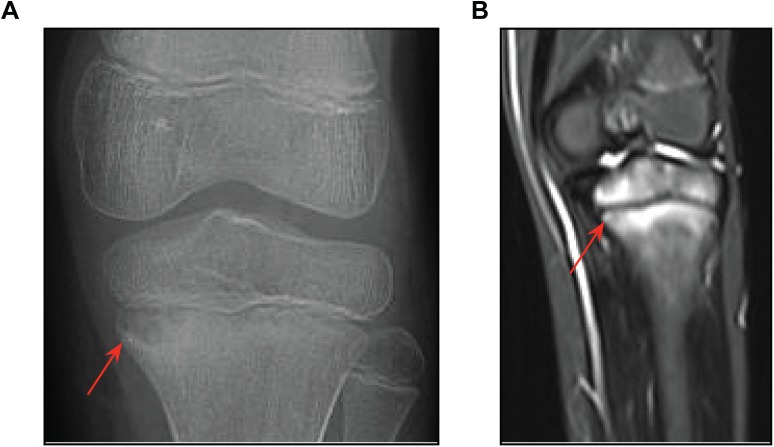
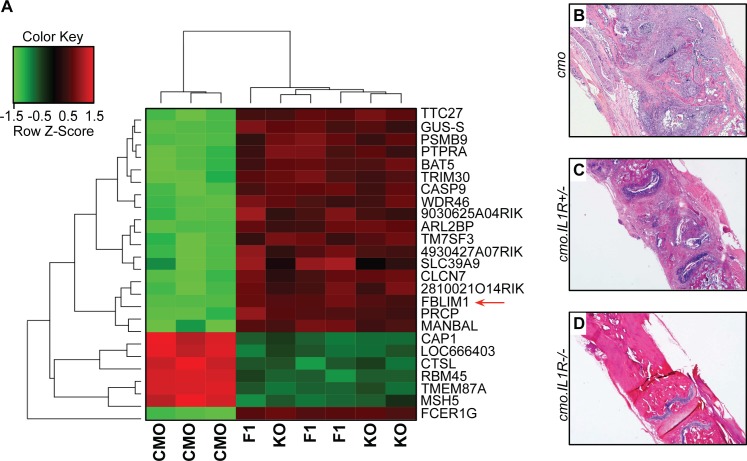
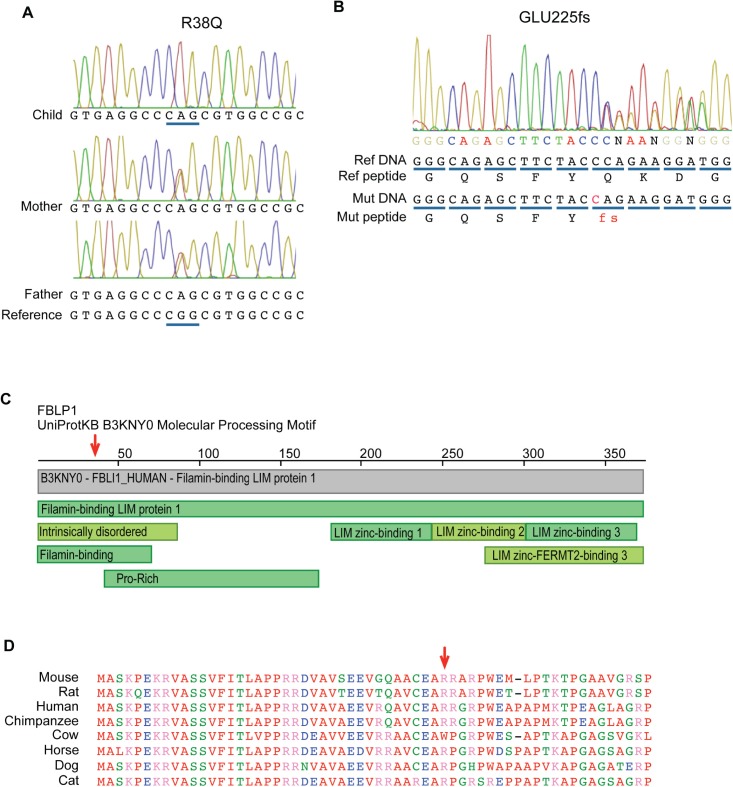
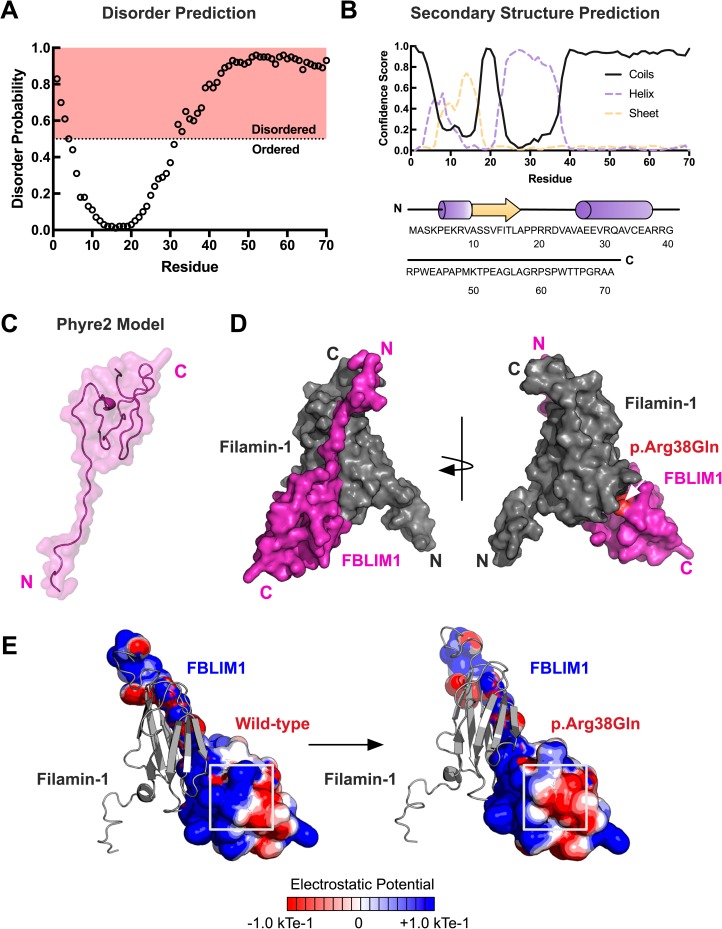
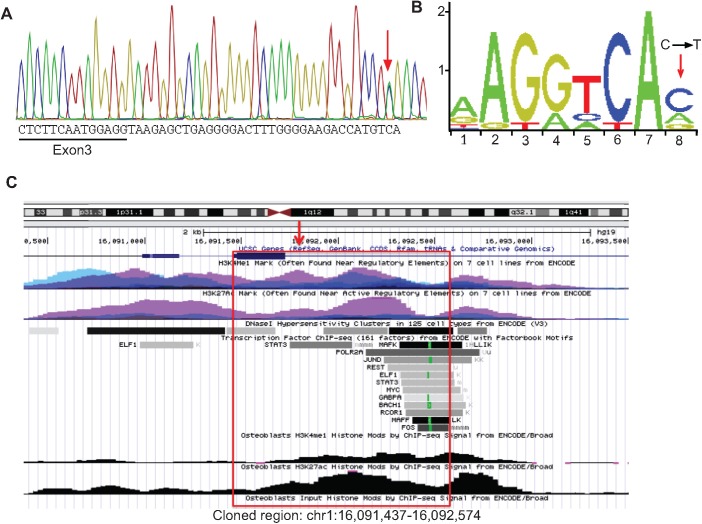
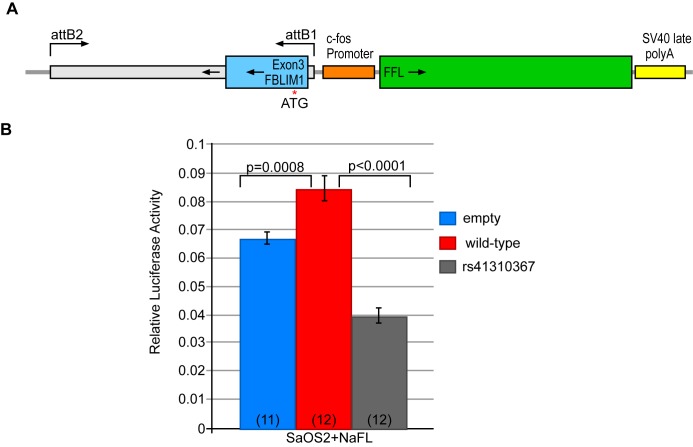
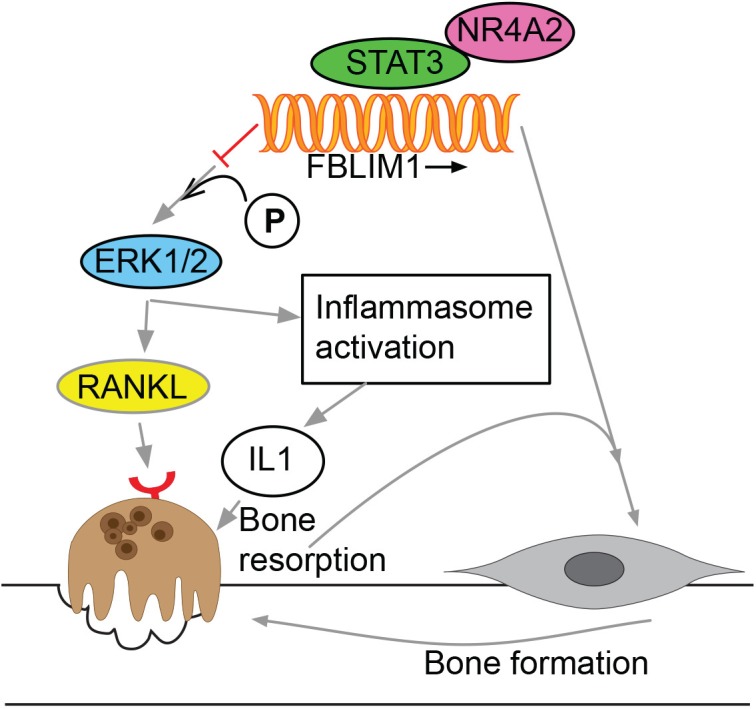
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare, pediatric, autoinflammatory disease characterized by bone pain due to sterile osteomyelitis, and is often accompanied by psoriasis or inflammatory bowel disease. There are two syndromic forms of CRMO, Majeed syndrome and DIRA, for which the genetic cause is known. However, for the majority of cases of CRMO, the genetic basis is unknown. Via whole-exome sequencing, we detected a homozygous mutation in the filamin-binding domain of FBLIM1 in an affected child with consanguineous parents. Microarray analysis of bone marrow macrophages from the CRMO murine model (cmo) determined that the Fblim1 ortholog is the most differentially expressed gene, downregulated over 20-fold in the cmo mouse. We sequenced FBLIM1 in 96 CRMO subjects and found a second proband with a novel frameshift mutation in exon 6 and a rare regulatory variant. In SaOS2 cells, overexpressing the regulatory mutation showed the flanking region acts as an enhancer, and the mutation ablates enhancer activity. Our data implicate FBLIM1 in the pathogenesis of sterile bone inflammation and our findings suggest CRMO is a disorder of chronic inflammation and imbalanced bone remodeling.
Conflict of interest statement
Figures
References
-
- Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). Journal of medical genetics. 2005;42(7):551–7. PubMed Central PMCID: PMC1736104. 10.1136/jmg.2005.030759 - DOI - PMC - PubMed
-
- Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. The New England journal of medicine. 2009;360(23):2426–37. PubMed Central PMCID: PMC2876877. 10.1056/NEJMoa0807865 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
