

Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability
- PMID: 28301528
- PMCID: PMC5354391
- DOI: 10.1371/journal.pone.0173723
Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability
Abstract
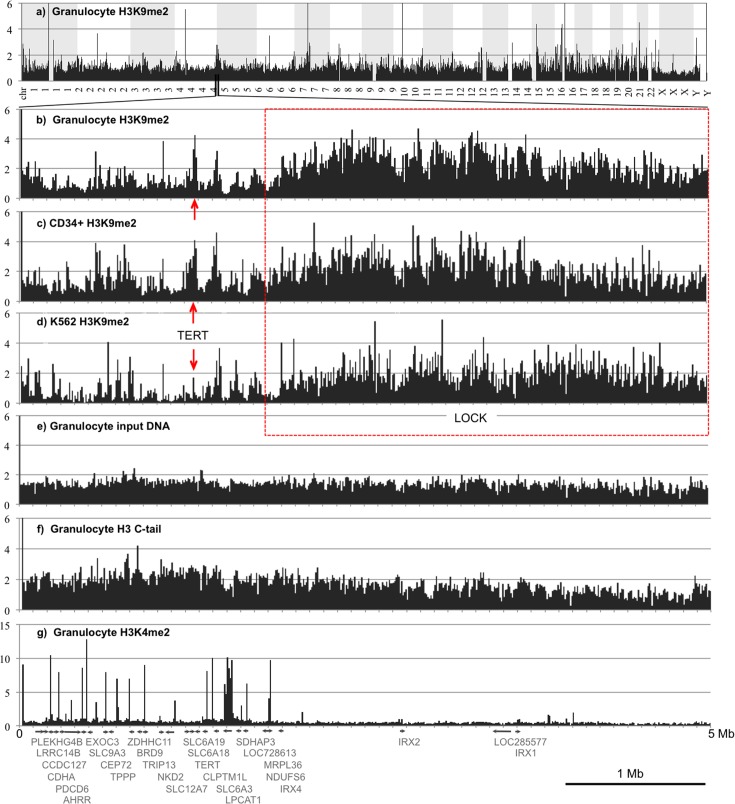
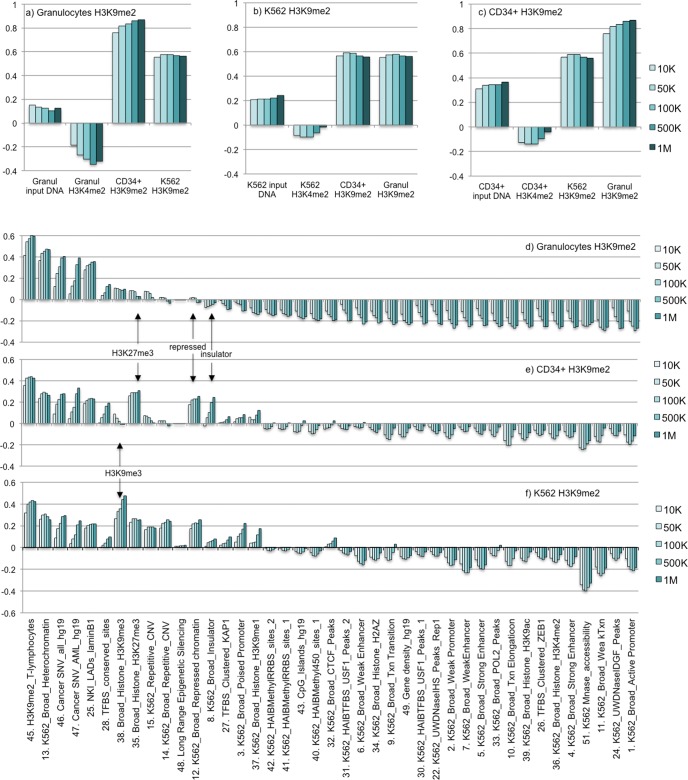
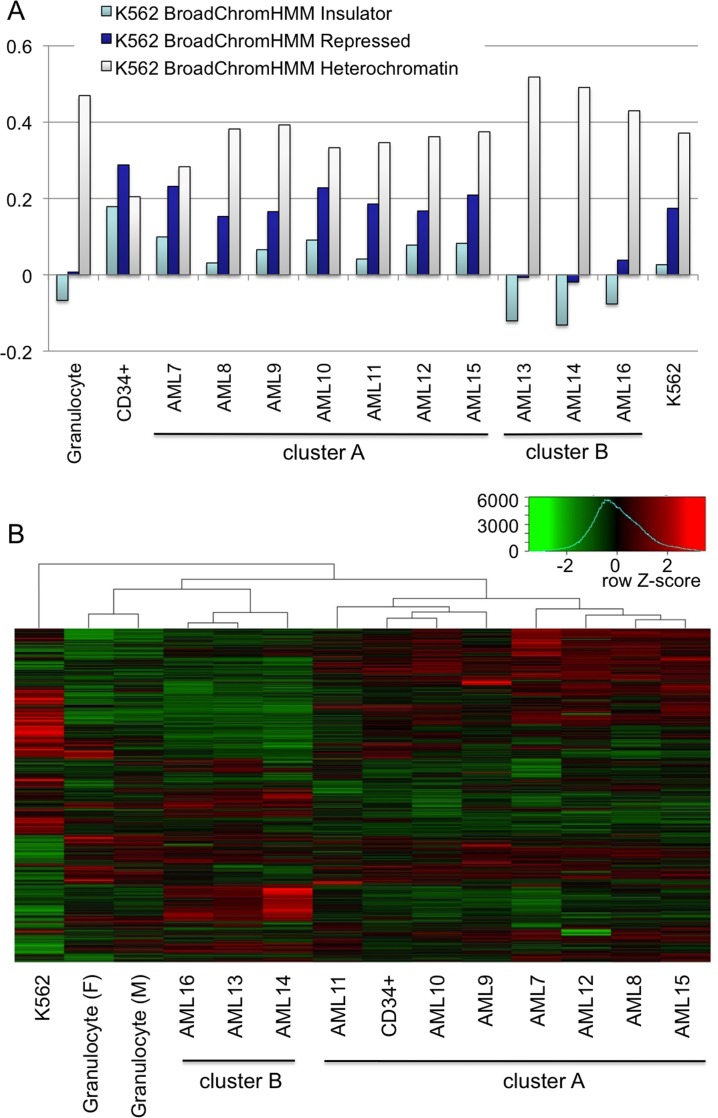
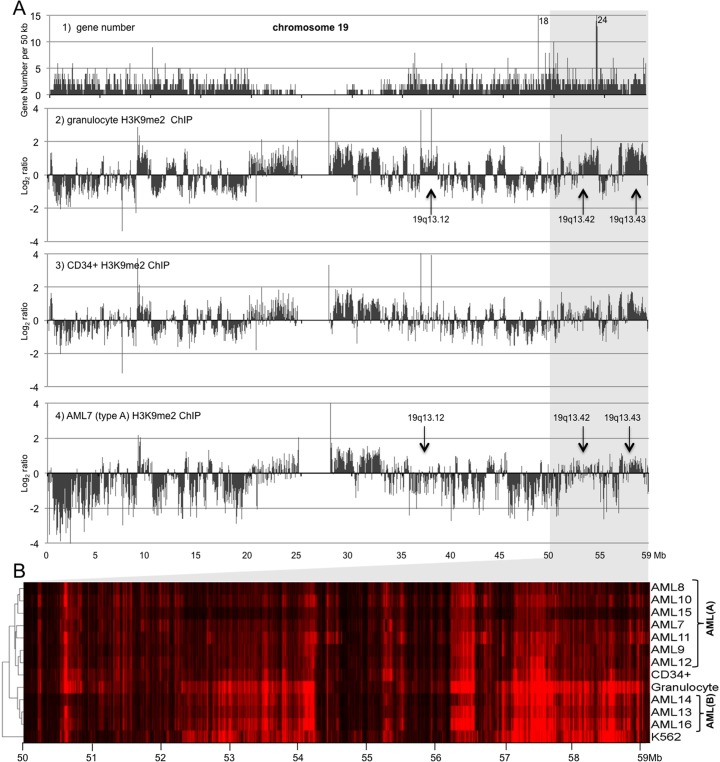
A facultative heterochromatin mark, histone H3 lysine 9 dimethylation (H3K9me2), which is mediated by histone methyltransferases G9a/GLP (EHMT2/1), undergoes dramatic rearrangements during myeloid cell differentiation as observed by chromatin imaging. To determine whether these structural transitions also involve genomic repositioning of H3K9me2, we used ChIP-sequencing to map genome-wide topography of H3K9me2 in normal human granulocytes, normal CD34+ hematopoietic progenitors, primary myeloblasts from acute myeloid leukemia (AML) patients, and a model leukemia cell line K562. We observe that H3K9me2 naturally repositions from the previously designated "repressed" chromatin state in hematopoietic progenitors to predominant association with heterochromatin regions in granulocytes. In contrast, AML cells accumulate H3K9me2 on previously undefined large (> 100 Kb) genomic blocks that are enriched with AML-specific single nucleotide variants, sites of chromosomal translocations, and genes downregulated in AML. Specifically, the AML-specific H3K9me2 blocks are enriched with genes regulated by the proto-oncogene ERG that promotes stem cell characteristics. The AML-enriched H3K9me2 blocks (in contrast to the heterochromatin-associated H3K9me2 blocks enriched in granulocytes) are reduced by pharmacological inhibition of the histone methyltransferase G9a/GLP in K562 cells concomitantly with transcriptional activation of ERG and ETS1 oncogenes. Our data suggest that G9a/GLP mediate formation of transient H3K9me2 blocks that are preserved in AML myeloblasts and may lead to an increased rate of AML-specific mutagenesis and chromosomal translocations.
Conflict of interest statement
Figures

References
-
- Kornberg RD, Lorch Y. Chromatin rules. Nat Struct Mol Biol. 2007;14(11):986–8.
-
- Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nature cell biology. 2013;15(7):872–82. Epub 2013/06/12. 10.1038/ncb2768 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
