

Is the Bethe-Salpeter Formalism Accurate for Excitation Energies? Comparisons with TD-DFT, CASPT2, and EOM-CCSD
- PMID: 28301726
- PMCID: PMC5385505
- DOI: 10.1021/acs.jpclett.7b00381
Is the Bethe-Salpeter Formalism Accurate for Excitation Energies? Comparisons with TD-DFT, CASPT2, and EOM-CCSD
Abstract
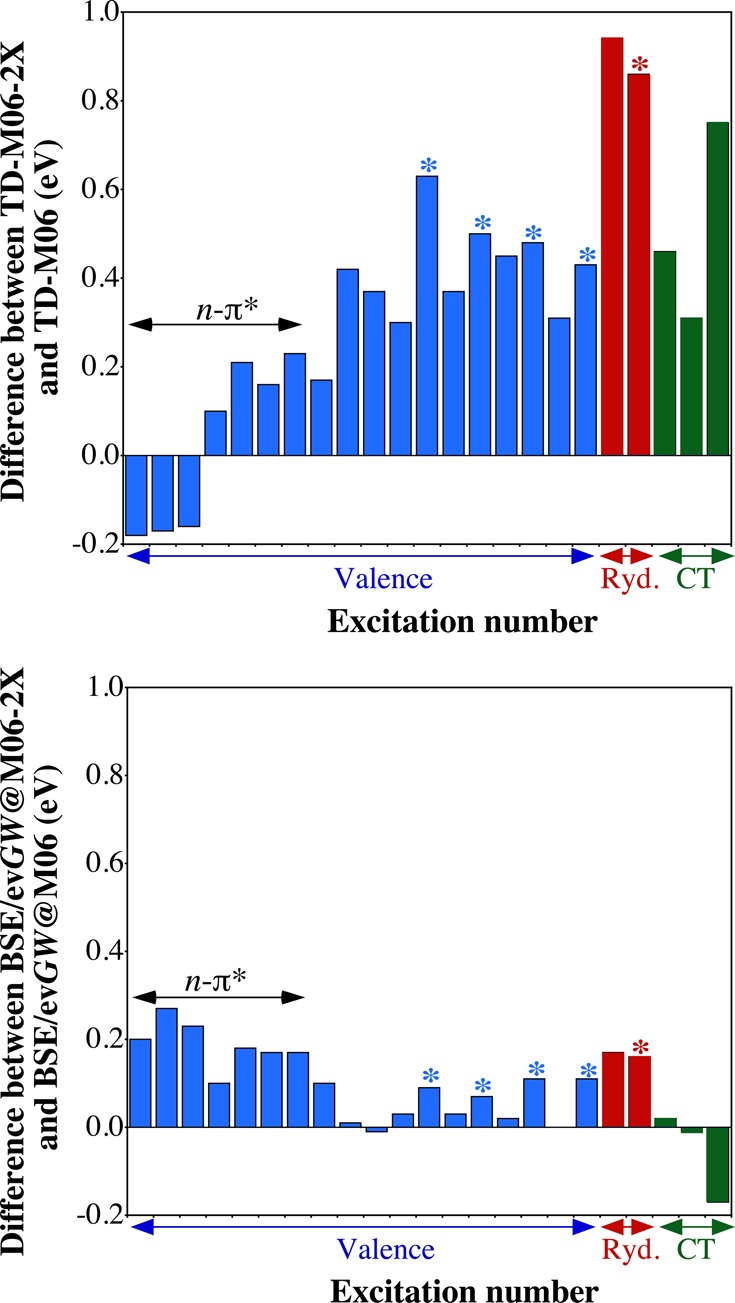
Developing ab initio approaches able to provide accurate excited-state energies at a reasonable computational cost is one of the biggest challenges in theoretical chemistry. In that framework, the Bethe-Salpeter equation approach, combined with the GW exchange-correlation self-energy, which maintains the same scaling with system size as TD-DFT, has recently been the focus of a rapidly increasing number of applications in molecular chemistry. Using a recently proposed set encompassing excitation energies of many kinds [J. Phys. Chem. Lett. 2016, 7, 586-591], we investigate here the performances of BSE/GW. We compare these results to CASPT2, EOM-CCSD, and TD-DFT data and show that BSE/GW provides an accuracy comparable to the two wave function methods. It is particularly remarkable that the BSE/GW is equally efficient for valence, Rydberg, and charge-transfer excitations. In contrast, it provides a poor description of triplet excited states, for which EOM-CCSD and CASPT2 clearly outperform BSE/GW. This contribution therefore supports the use of the Bethe-Salpeter approach for spin-conserving transitions.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Ullrich C.Time-Dependent Density-Functional Theory: Concepts and Applications; Oxford Graduate Texts; Oxford University Press: New York, 2012.
-
- Santoro F.; Jacquemin D. Going Beyond the Vertical Approximation with Time-Dependent Density Functional Theory. WIREs Comput. Mol. Sci. 2016, 6, 460–486. 10.1002/wcms.1260. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
