

Biophysical Models of Protein Evolution: Understanding the Patterns of Evolutionary Sequence Divergence
- PMID: 28301766
- PMCID: PMC5800964
- DOI: 10.1146/annurev-biophys-070816-033819
Biophysical Models of Protein Evolution: Understanding the Patterns of Evolutionary Sequence Divergence
Abstract
For decades, rates of protein evolution have been interpreted in terms of the vague concept of functional importance. Slowly evolving proteins or sites within proteins were assumed to be more functionally important and thus subject to stronger selection pressure. More recently, biophysical models of protein evolution, which combine evolutionary theory with protein biophysics, have completely revolutionized our view of the forces that shape sequence divergence. Slowly evolving proteins have been found to evolve slowly because of selection against toxic misfolding and misinteractions, linking their rate of evolution primarily to their abundance. Similarly, most slowly evolving sites in proteins are not directly involved in function, but mutating these sites has a large impact on protein structure and stability. In this article, we review the studies in the emerging field of biophysical protein evolution that have shaped our current understanding of sequence divergence patterns. We also propose future research directions to develop this nascent field.
Keywords: evolutionary rate; fitness landscape; protein folding; protein misfolding; protein–protein interaction.
Figures
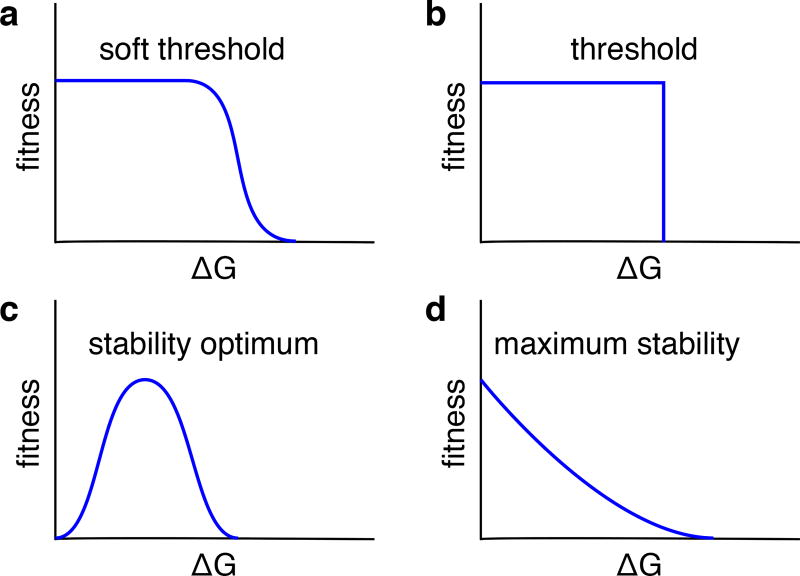
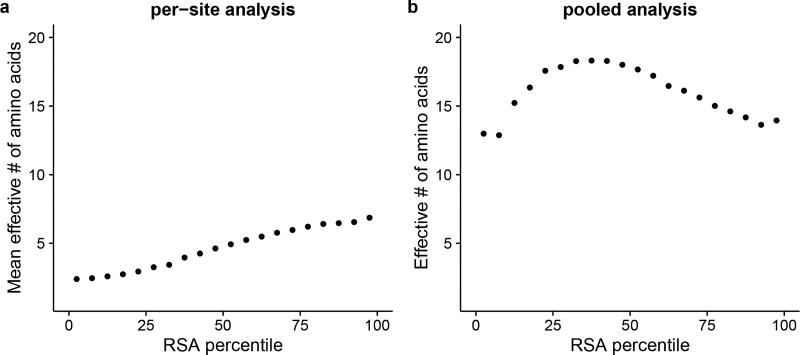
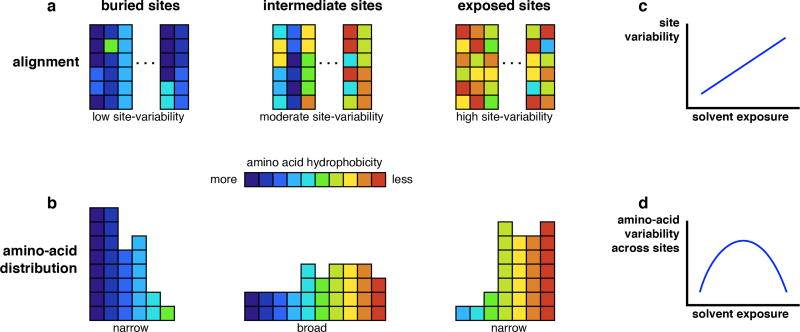
References
-
- Bastolla U, Porto M, Roman HE, Vendruscolo M. Principal eigenvector of contact matrices and hydrophobicity profiles in proteins. Proteins. 2004;58:22–30. - PubMed
-
- Bloom JD, Drummond DA, Arnold FH, Wilke CO. Structural determinants of the rate of protein evolution in yeast. Mol. Biol. Evol. 2006;23:1751–1761. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
