

Relationship of acute axonal damage, Wallerian degeneration, and clinical disability in multiple sclerosis
- PMID: 28302146
- PMCID: PMC5356322
- DOI: 10.1186/s12974-017-0831-8
Relationship of acute axonal damage, Wallerian degeneration, and clinical disability in multiple sclerosis
Abstract
Background: Axonal damage and loss substantially contribute to the incremental accumulation of clinical disability in progressive multiple sclerosis. Here, we assessed the amount of Wallerian degeneration in brain tissue of multiple sclerosis patients in relation to demyelinating lesion activity and asked whether a transient blockade of Wallerian degeneration decreases axonal loss and clinical disability in a mouse model of inflammatory demyelination.
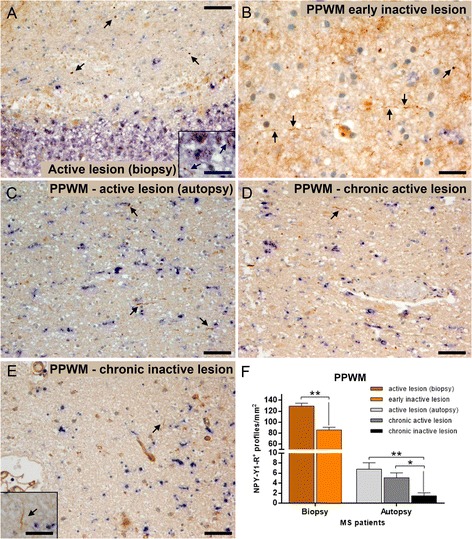
Methods: Wallerian degeneration and acute axonal damage were determined immunohistochemically in the periplaque white matter of multiple sclerosis patients with early actively demyelinating lesions, chronic active lesions, and inactive lesions. Furthermore, we studied the effects of Wallerian degeneration blockage on clinical severity, inflammatory pathology, acute axonal damage, and long-term axonal loss in experimental autoimmune encephalomyelitis using Wallerian degeneration slow (Wld S ) mutant mice.
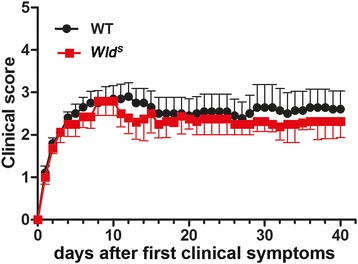
Results: The highest numbers of axons undergoing Wallerian degeneration were found in the perilesional white matter of multiple sclerosis patients early in the disease course and with actively demyelinating lesions. Furthermore, Wallerian degeneration was more abundant in patients harboring chronic active as compared to chronic inactive lesions. No co-localization of neuropeptide Y-Y1 receptor, a bona fide immunohistochemical marker of Wallerian degeneration, with amyloid precursor protein, frequently used as an indicator of acute axonal transport disturbance, was observed in human and mouse tissue, indicating distinct axon-degenerative processes. Experimentally, a delay of Wallerian degeneration, as observed in Wld S mice, did not result in a reduction of clinical disability or acute axonal damage in experimental autoimmune encephalomyelitis, further supporting that acute axonal damage as reflected by axonal transport disturbances does not share common molecular mechanisms with Wallerian degeneration. Furthermore, delaying Wallerian degeneration did not result in a net rescue of axons in late lesion stages of experimental autoimmune encephalomyelitis.
Conclusions: Our data indicate that in multiple sclerosis, ongoing demyelination in focal lesions is associated with axonal degeneration in the perilesional white matter, supporting a role for focal pathology in diffuse white matter damage. Also, our results suggest that interfering with Wallerian degeneration in inflammatory demyelination does not suffice to prevent acute axonal damage and finally axonal loss.
Keywords: Axonal damage; Experimental autoimmune encephalomyelitis; Multiple sclerosis; Wallerian degeneration; Wld S.
Figures




Similar articles
-
Wallerian degeneration: a major component of early axonal pathology in multiple sclerosis.Brain Pathol. 2010 Sep;20(5):976-85. doi: 10.1111/j.1750-3639.2010.00401.x. Epub 2010 Apr 14. Brain Pathol. 2010. PMID: 20477831 Free PMC article.
-
Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions.Am J Pathol. 2000 Jul;157(1):267-76. doi: 10.1016/S0002-9440(10)64537-3. Am J Pathol. 2000. PMID: 10880396 Free PMC article.
-
Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation.Brain. 2012 Jun;135(Pt 6):1794-818. doi: 10.1093/brain/aws100. Epub 2012 Apr 28. Brain. 2012. PMID: 22544872 Free PMC article.
-
Neuroprotective strategies in MS: lessons from C57BL/Wld(S) mice.J Neurol Sci. 2005 Jun 15;233(1-2):133-8. doi: 10.1016/j.jns.2005.03.028. J Neurol Sci. 2005. PMID: 15899498 Review.
-
Inflammatory demyelination is not central to the pathogenesis of multiple sclerosis.J Neurol. 2005 Nov;252 Suppl 5:v10-5. doi: 10.1007/s00415-005-5003-6. J Neurol. 2005. PMID: 16254696 Review.
Cited by
-
Animal models to investigate the effects of inflammation on remyelination in multiple sclerosis.Front Mol Neurosci. 2022 Nov 3;15:995477. doi: 10.3389/fnmol.2022.995477. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36407761 Free PMC article. Review.
-
Tract-wise microstructural analysis informs on current and future disability in early multiple sclerosis.J Neurol. 2024 Feb;271(2):631-641. doi: 10.1007/s00415-023-12023-3. Epub 2023 Oct 11. J Neurol. 2024. PMID: 37819462 Free PMC article.
-
Chronic White Matter Inflammation and Serum Neurofilament Levels in Multiple Sclerosis.Neurology. 2021 Aug 10;97(6):e543-e553. doi: 10.1212/WNL.0000000000012326. Epub 2021 Jun 4. Neurology. 2021. PMID: 34088875 Free PMC article.
-
The Distributional Characteristics of Multiple Sclerosis Lesions on Quantitative Susceptibility Mapping and Their Correlation With Clinical Severity.Front Neurol. 2021 Jul 9;12:647519. doi: 10.3389/fneur.2021.647519. eCollection 2021. Front Neurol. 2021. PMID: 34305779 Free PMC article.
-
Brief electrical nerve stimulation enhances intrinsic repair capacity of the focally demyelinated central nervous system.Neural Regen Res. 2022 May;17(5):1042-1050. doi: 10.4103/1673-5374.324848. Neural Regen Res. 2022. PMID: 34558531 Free PMC article.
References
-
- Losseff NA, Webb SL, O'Riordan JI, Page R, Wang L, Barker GJ, Tofts PS, McDonald WI, Miller DH, Thompson AJ. Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain. 1996;119(Pt 3):701–708. doi: 10.1093/brain/119.3.701. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
