

PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity
- PMID: 28302645
- PMCID: PMC5379970
- DOI: 10.1084/jem.20160801
PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity
Abstract
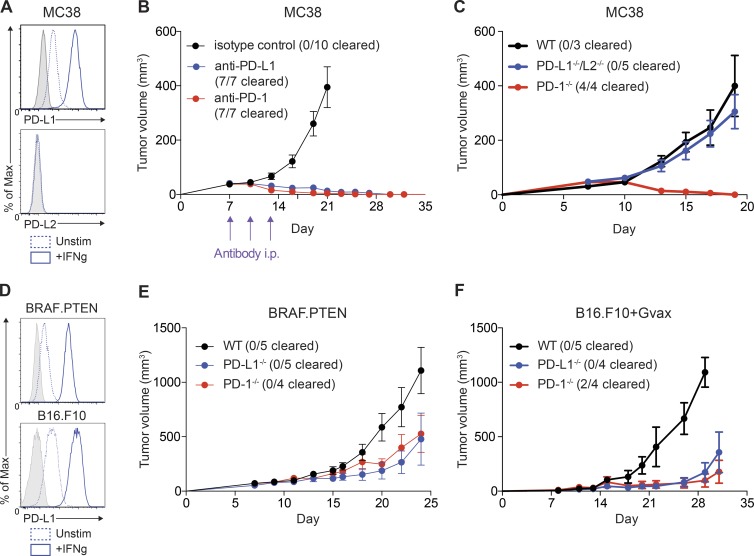
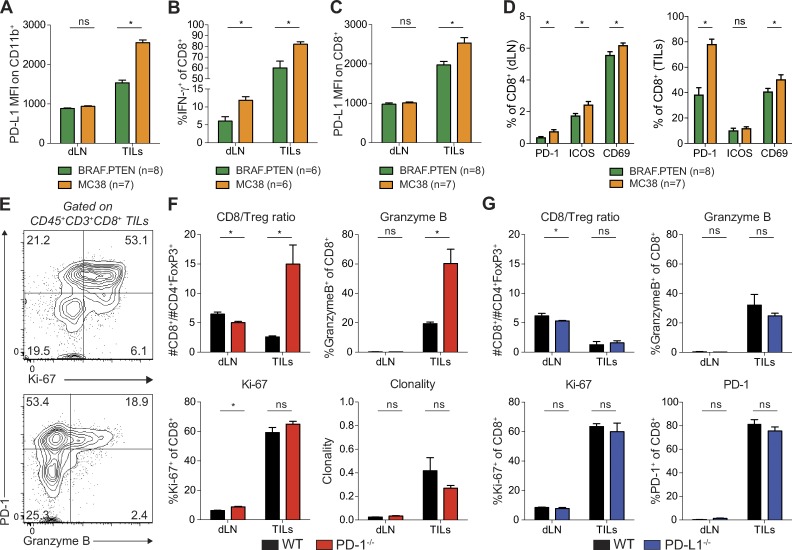
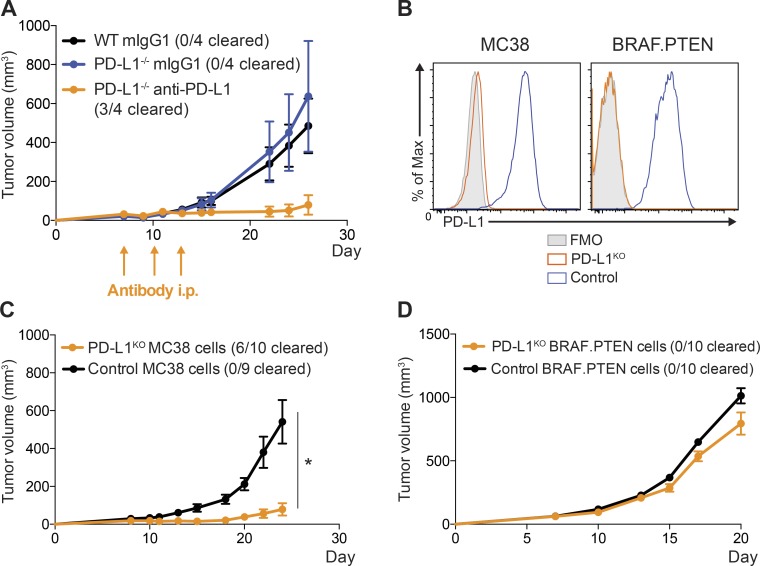
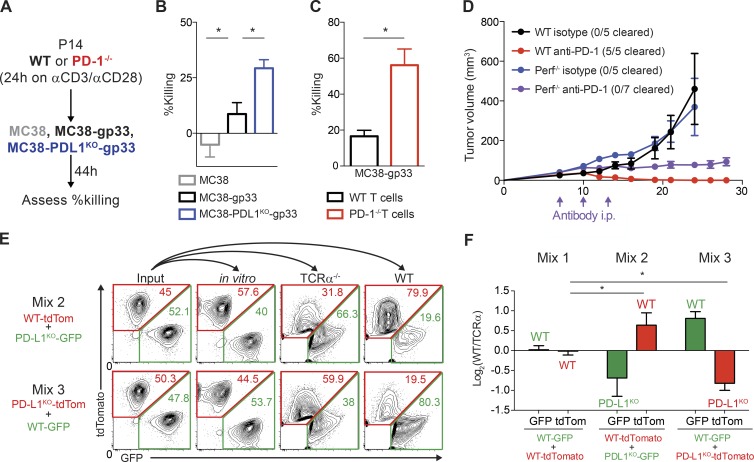
It is unclear whether PD-L1 on tumor cells is sufficient for tumor immune evasion or simply correlates with an inflamed tumor microenvironment. We used three mouse tumor models sensitive to PD-1 blockade to evaluate the significance of PD-L1 on tumor versus nontumor cells. PD-L1 on nontumor cells is critical for inhibiting antitumor immunity in B16 melanoma and a genetically engineered melanoma. In contrast, PD-L1 on MC38 colorectal adenocarcinoma cells is sufficient to suppress antitumor immunity, as deletion of PD-L1 on highly immunogenic MC38 tumor cells allows effective antitumor immunity. MC38-derived PD-L1 potently inhibited CD8+ T cell cytotoxicity. Wild-type MC38 cells outcompeted PD-L1-deleted MC38 cells in vivo, demonstrating tumor PD-L1 confers a selective advantage. Thus, both tumor- and host-derived PD-L1 can play critical roles in immunosuppression. Differences in tumor immunogenicity appear to underlie their relative importance. Our findings establish reduced cytotoxicity as a key mechanism by which tumor PD-L1 suppresses antitumor immunity and demonstrate that tumor PD-L1 is not just a marker of suppressed antitumor immunity.
© 2017 Juneja et al.
Figures
References
-
- Akhmetzyanova I., Drabczyk M., Neff C.P., Gibbert K., Dietze K.K., Werner T., Liu J., Chen L., Lang K.S., Palmer B.E., et al. . 2015. PD-L1 expression on retrovirus-infected cells mediates immune escape from CD8+ T cell killing. PLoS Pathog. 11:e1005224 10.1371/journal.ppat.1005224 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
