

Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma
- PMID: 28302680
- PMCID: PMC5413372
- DOI: 10.1158/0008-5472.CAN-16-2822
Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma
Abstract
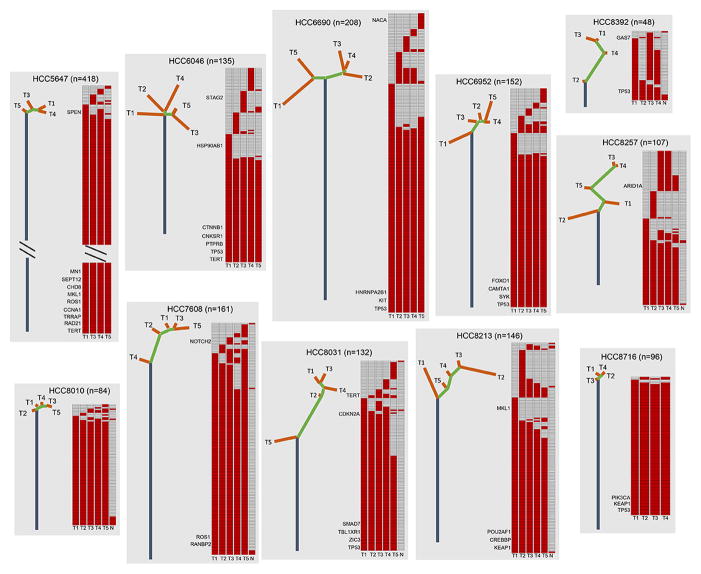
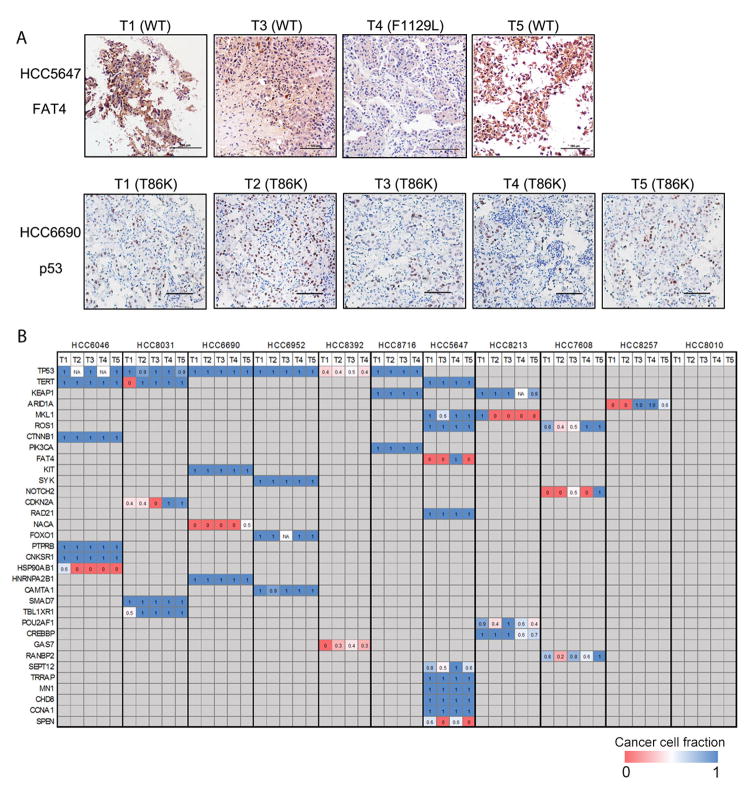
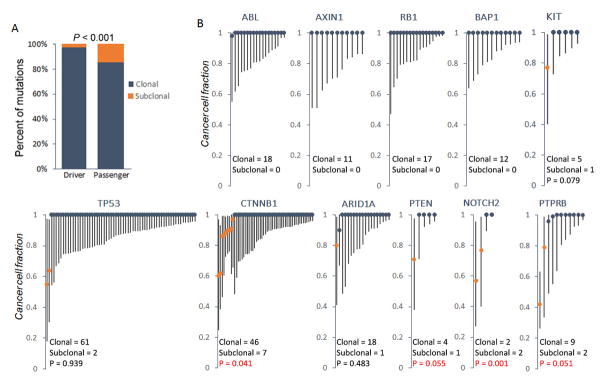
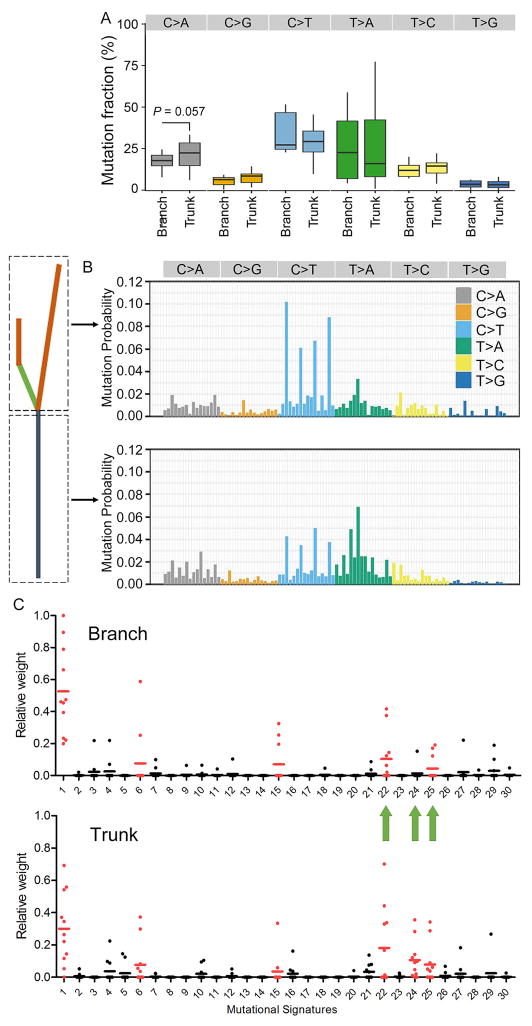
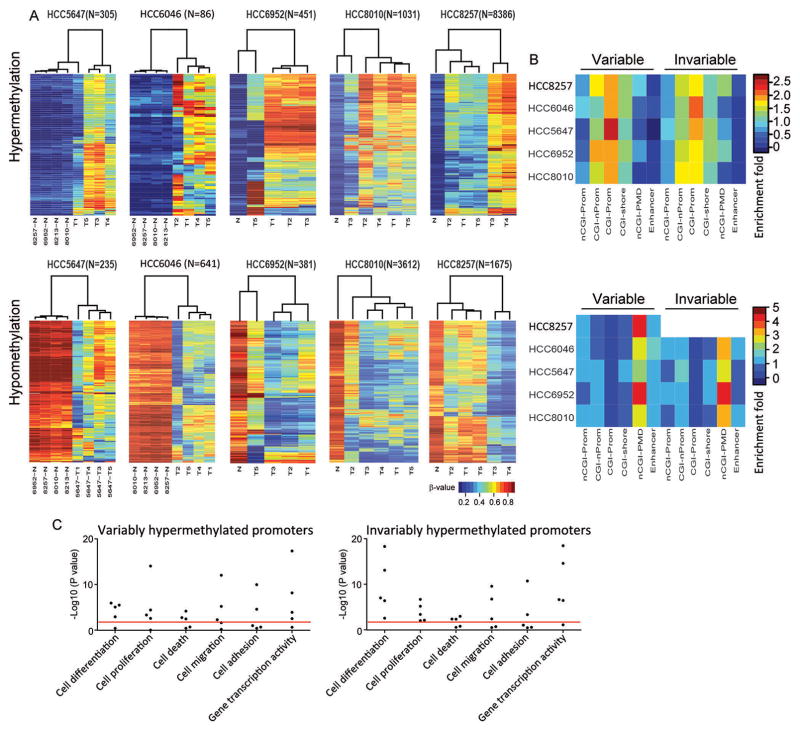
Understanding the intratumoral heterogeneity of hepatocellular carcinoma is instructive for developing personalized therapy and identifying molecular biomarkers. Here we applied whole-exome sequencing to 69 samples from 11 patients to resolve the genetic architecture of subclonal diversification. Spatial genomic diversity was found in all 11 hepatocellular carcinoma cases, with 29% of driver mutations being heterogeneous, including TERT, ARID1A, NOTCH2, and STAG2. Similar with other cancer types, TP53 mutations were always shared between all tumor regions, that is, located on the "trunk" of the evolutionary tree. In addition, we found that variants within several drug targets such as KIT, SYK, and PIK3CA were mutated in a fully clonal manner, indicating their therapeutic potentials for hepatocellular carcinoma. Temporal dissection of mutational signatures suggested that mutagenic processes associated with exposure to aristolochic acid and aflatoxin might play a more important role in early, as opposed to late, stages of hepatocellular carcinoma development. Moreover, we observed extensive intratumoral epigenetic heterogeneity in hepatocellular carcinoma based on multiple independent analytical methods and showed that intratumoral methylation heterogeneity might play important roles in the biology of hepatocellular carcinoma cells. Our results also demonstrated prominent heterogeneity of intratumoral methylation even in a stable hepatocellular carcinoma genome. Together, these findings highlight widespread intratumoral heterogeneity at both the genomic and epigenomic levels in hepatocellular carcinoma and provide an important molecular foundation for better understanding the pathogenesis of this malignancy. Cancer Res; 77(9); 2255-65. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
The authors have no potential conflicts of interest to disclose.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
