

Immune checkpoint dysfunction in large and medium vessel vasculitis
- PMID: 28314758
- PMCID: PMC5451585
- DOI: 10.1152/ajpheart.00024.2017
Immune checkpoint dysfunction in large and medium vessel vasculitis
Abstract
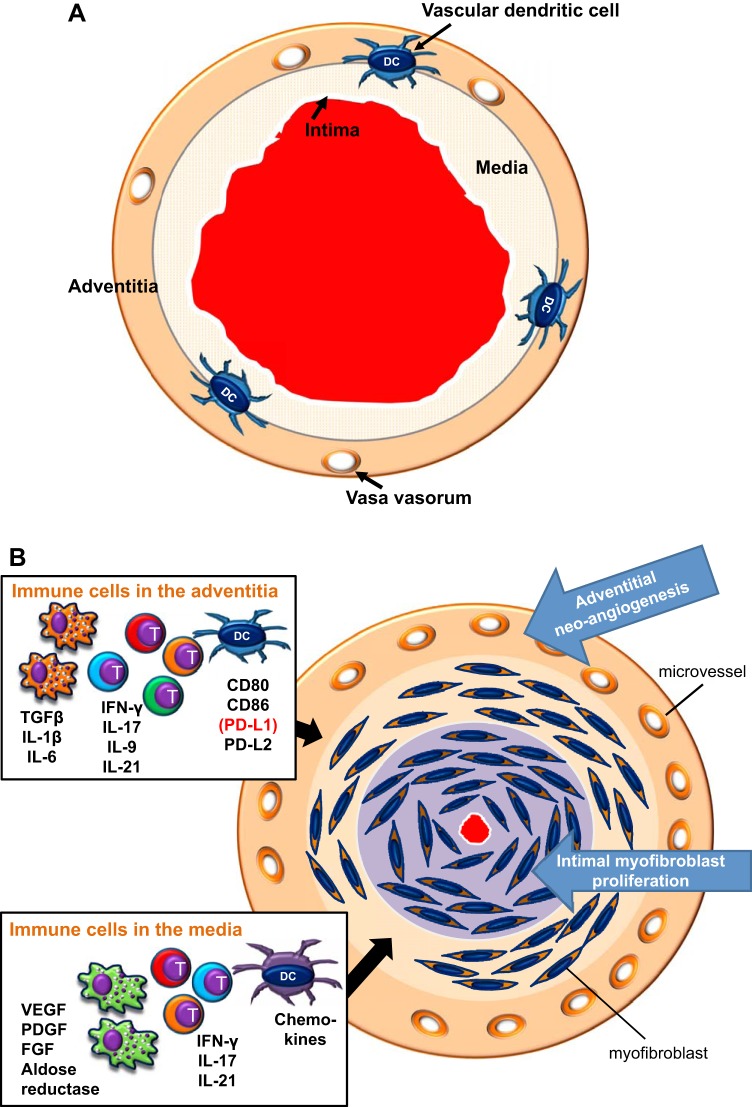
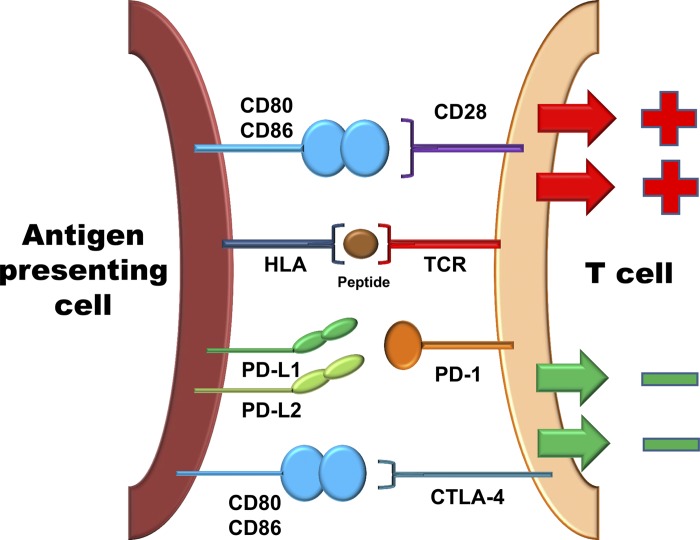
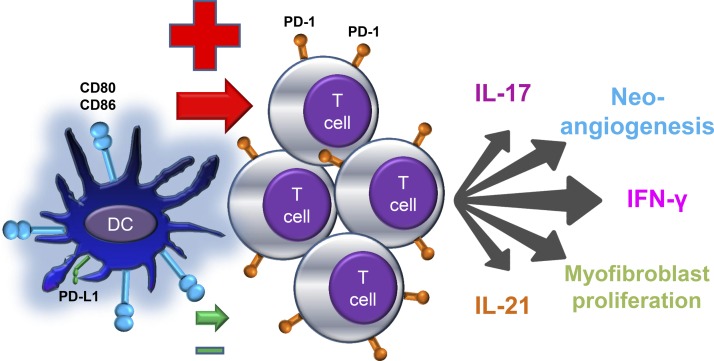
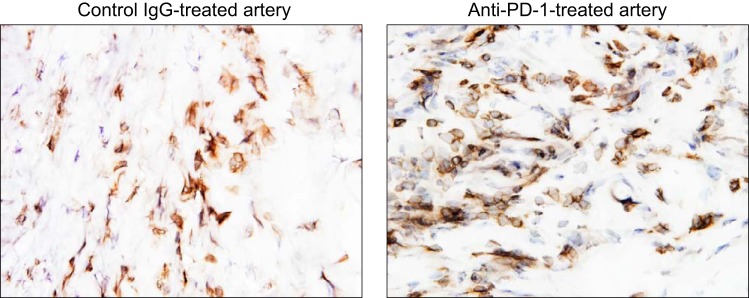
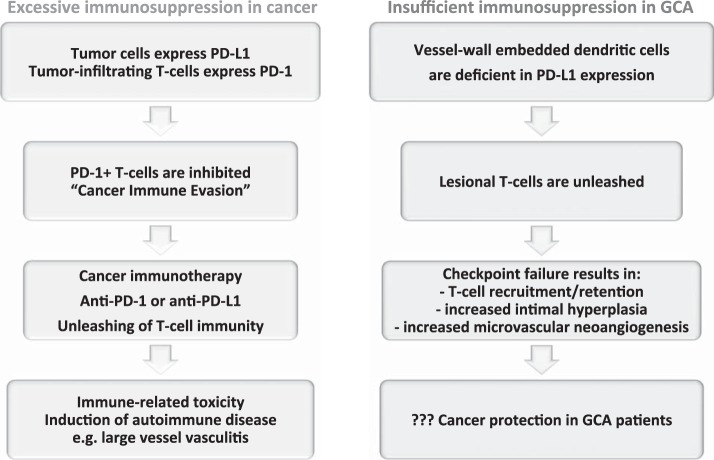
Giant cell arteritis (GCA) is a granulomatous vasculitis of the aorta and its medium-sized branch vessels. CD4 T cells, macrophages, and dendritic cells (DCs) build granulomatous infiltrates that injure the vessel wall and elicit a maladaptive response to injury. Pathological consequences include fragmentation of elastic membranes, destruction of the medial layer, microvascular neoangiogenesis, massive outgrowth of myofibroblasts, and lumen-occlusive intimal hyperplasia. Antigens have been suspected to drive the local activation of vasculitogenic CD4 T cells, but recent data have suggested a more generalized defect in the threshold setting of such T cells, rendering them hyperreactive. Under physiological conditions, immune checkpoints provide negative signals to curb T cell activation and prevent inflammation-associated tissue destruction. This protective mechanism is disrupted in GCA. Vessel wall DCs fail to express the immunoinhibitory ligand programmed cell death ligand-1, leaving lesional T cells unchecked. Consequently, programmed cell death protein-1-positive CD4 T cells can enter the immunoprivileged vessel wall, where they produce a broad spectrum of inflammatory cytokines (interferon-γ, IL-17, and IL-21) and have a direct role in driving intimal hyperplasia and intramural neoangiogenesis. The deficiency of the programmed cell death protein-1 immune checkpoint in GCA, promoting unopposed T cell immunity, contrasts with checkpoint hyperactivity in cancer patients in whom excessive programmed cell death ligand-1 expression paralyzes the function of antitumor T cells. Excessive checkpoint activity is the principle underlying cancer-immune evasion and is therapeutically targeted by immunotherapy with checkpoint inhibitors. Such checkpoint inhibitors, which unleash anticancer T cells and induce immune-related toxicity, may lead to drug-induced vasculitis.
Keywords: dendritic cell; giant cell arteritis; immune checkpoint; programmed cell death ligand-1; programmed cell death protein-1.
Copyright © 2017 the American Physiological Society.
Figures
References
-
- Bu DX, Tarrio M, Maganto-Garcia E, Stavrakis G, Tajima G, Lederer J, Jarolim P, Freeman GJ, Sharpe AH, Lichtman AH. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol 31: 1100–1107, 2011. doi: 10.1161/ATVBAHA.111.224709. - DOI - PMC - PubMed
-
- Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, Cannizzaro A, Boiardi L, Iacopino DG, Salvarani C, Triolo G. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology (Oxford) 54: 1596–1604, 2015. doi: 10.1093/rheumatology/kev102. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials