

TRPM4 non-selective cation channel variants in long QT syndrome
- PMID: 28315637
- PMCID: PMC5357330
- DOI: 10.1186/s12881-017-0397-4
TRPM4 non-selective cation channel variants in long QT syndrome
Abstract
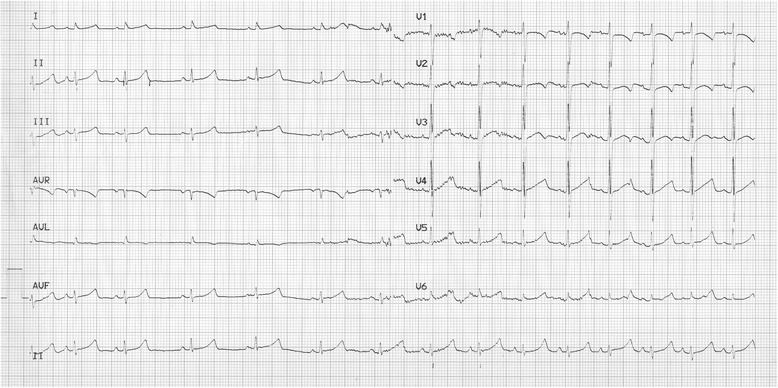
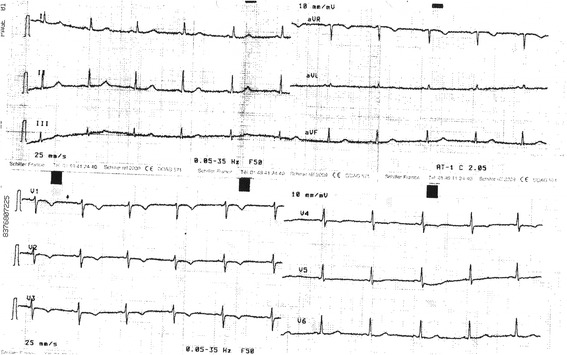
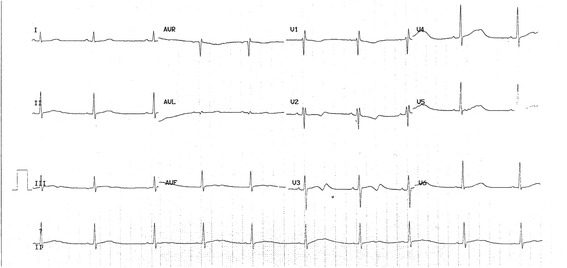
Background: Long QT syndrome (LQTS) is an inherited arrhythmic disorder characterized by prolongation of the QT interval, a risk of syncope, and sudden death. There are already a number of causal genes in LQTS, but not all LQTS patients have an identified mutation, which suggests LQTS unknown genes.
Methods: A cohort of 178 LQTS patients, with no mutations in the 3 major LQTS genes (KCNQ1, KCNH2, and SCN5A), was screened for mutations in the transient potential melastatin 4 gene (TRPM4).
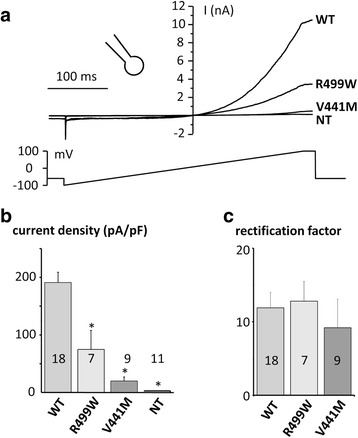
Results: Four TRPM4 variants (2.2% of the cohort) were found to change highly conserved amino-acids and were either very rare or absent from control populations. Therefore, these four TRPM4 variants were predicted to be disease causing. Furthermore, no mutations were found in the DNA of these TRPM4 variant carriers in any of the 13 major long QT syndrome genes. Two of these variants were further studied by electrophysiology (p.Val441Met and p.Arg499Pro). Both variants showed a classical TRPM4 outward rectifying current, but the current was reduced by 61 and 90% respectively, compared to wild type TRPM4 current.
Conclusions: This study supports the view that TRPM4 could account for a small percentage of LQTS patients. TRPM4 contribution to the QT interval might be multifactorial by modulating whole cell current but also, as shown in Trpm4-/- mice, by modulating cardiomyocyte proliferation. TRPM4 enlarges the subgroup of LQT genes (KCNJ2 in Andersen syndrome and CACNA1C in Timothy syndrome) known to increase the QT interval through a more complex pleiotropic effect than merely action potential alteration.
Keywords: Arrhythmia; Electrophysiology; Gene mutation; Long QT syndrome; Outward rectifying current; TRPM4.
Figures


References
-
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–39. doi: 10.1016/j.hrthm.2011.05.020. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
