

Computational methods for prediction of in vitro effects of new chemical structures
- PMID: 28316649
- PMCID: PMC5043617
- DOI: 10.1186/s13321-016-0162-2
Computational methods for prediction of in vitro effects of new chemical structures
Abstract
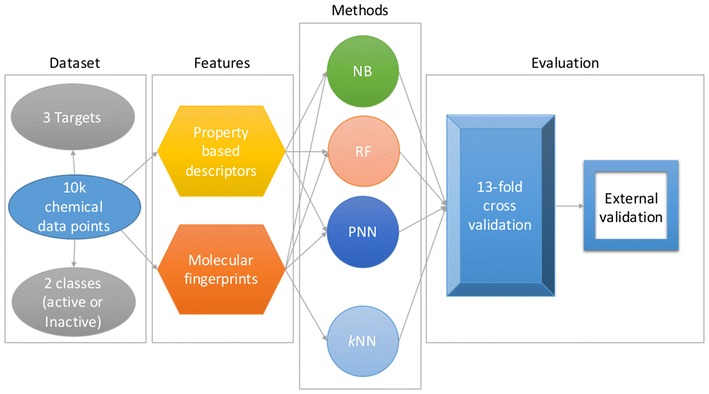
Background: With a constant increase in the number of new chemicals synthesized every year, it becomes important to employ the most reliable and fast in silico screening methods to predict their safety and activity profiles. In recent years, in silico prediction methods received great attention in an attempt to reduce animal experiments for the evaluation of various toxicological endpoints, complementing the theme of replace, reduce and refine. Various computational approaches have been proposed for the prediction of compound toxicity ranging from quantitative structure activity relationship modeling to molecular similarity-based methods and machine learning. Within the "Toxicology in the 21st Century" screening initiative, a crowd-sourcing platform was established for the development and validation of computational models to predict the interference of chemical compounds with nuclear receptor and stress response pathways based on a training set containing more than 10,000 compounds tested in high-throughput screening assays.
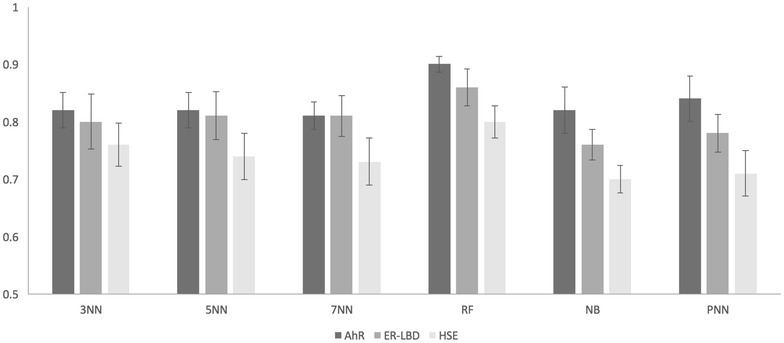
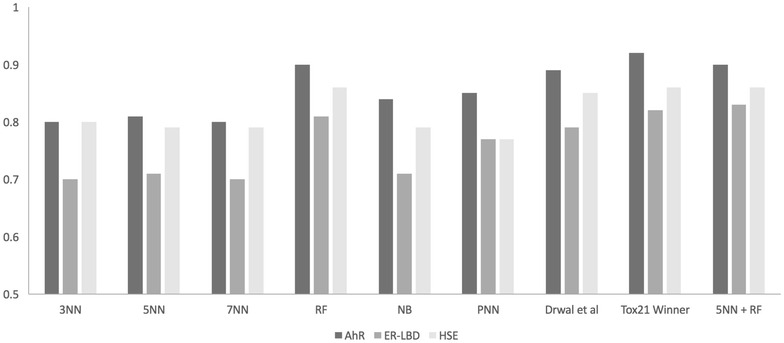
Results: Here, we present the results of various molecular similarity-based and machine-learning based methods over an independent evaluation set containing 647 compounds as provided by the Tox21 Data Challenge 2014. It was observed that the Random Forest approach based on MACCS molecular fingerprints and a subset of 13 molecular descriptors selected based on statistical and literature analysis performed best in terms of the area under the receiver operating characteristic curve values. Further, we compared the individual and combined performance of different methods. In retrospect, we also discuss the reasons behind the superior performance of an ensemble approach, combining a similarity search method with the Random Forest algorithm, compared to individual methods while explaining the intrinsic limitations of the latter.
Conclusions: Our results suggest that, although prediction methods were optimized individually for each modelled target, an ensemble of similarity and machine-learning approaches provides promising performance indicating its broad applicability in toxicity prediction.
Keywords: Machine learning; Molecular fingerprints; Similarity searching; Tox21 challenge; Toxicity prediction.
Figures
References
-
- Vedani A, Smiesko M. In silico toxicology in drug discovery—concepts based on three-dimensional models. Altern Lab Anim ATLA. 2009;37:477–496. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
