

Insights into molecular therapy of glioma: current challenges and next generation blueprint
- PMID: 28317871
- PMCID: PMC5457688
- DOI: 10.1038/aps.2016.167
Insights into molecular therapy of glioma: current challenges and next generation blueprint
Abstract
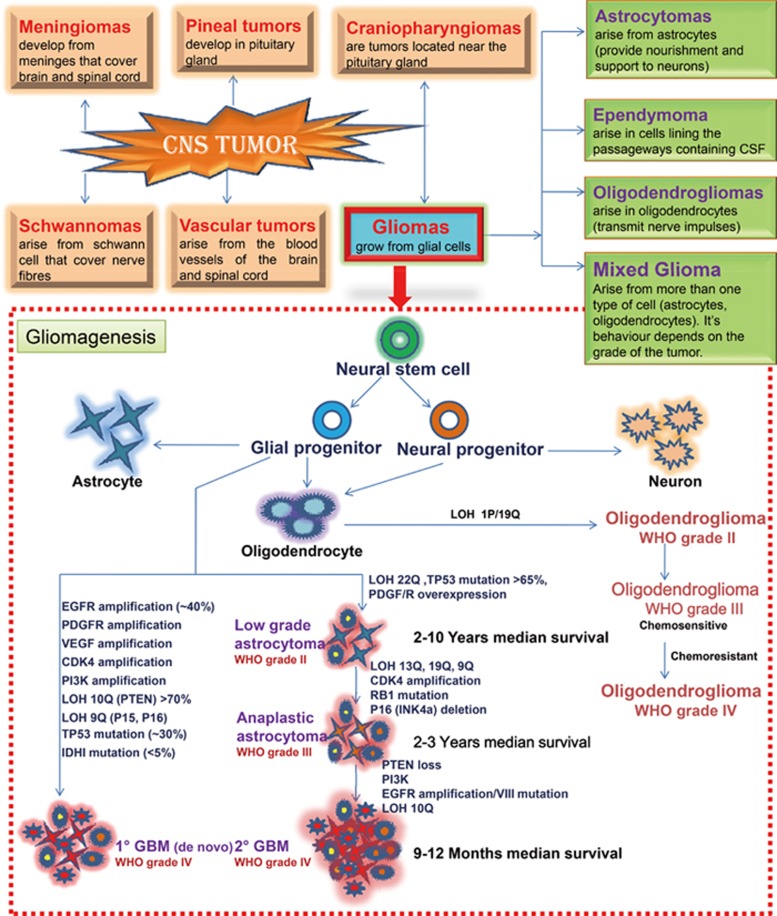
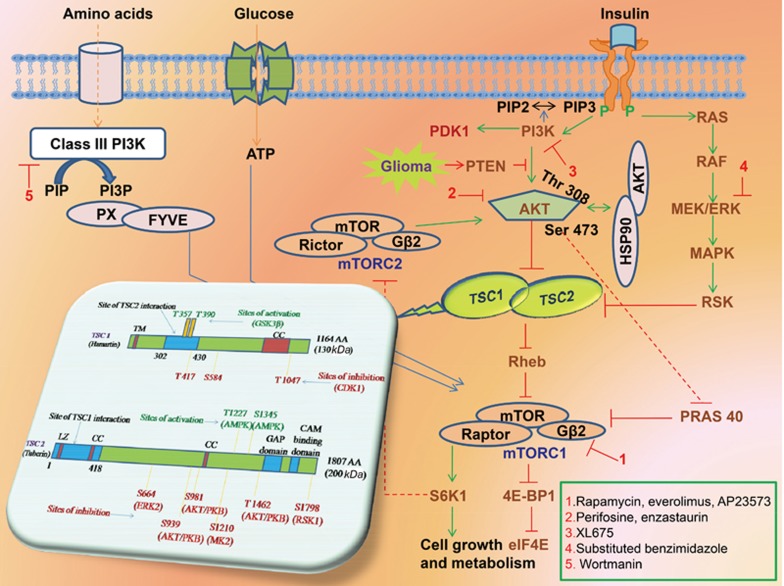
Glioma accounts for the majority of human brain tumors. With prevailing treatment regimens, the patients have poor survival rates. In spite of current development in mainstream glioma therapy, a cure for glioma appears to be out of reach. The infiltrative nature of glioma and acquired resistance substancially restrict the therapeutic options. Better elucidation of the complicated pathobiology of glioma and proteogenomic characterization might eventually open novel avenues for the design of more sophisticated and effective combination regimens. This could be accomplished by individually tailoring progressive neuroimaging techniques, terminating DNA synthesis with prodrug-activating genes, silencing gliomagenesis genes (gene therapy), targeting miRNA oncogenic activity (miRNA-mRNA interaction), combining Hedgehog-Gli/Akt inhibitors with stem cell therapy, employing tumor lysates as antigen sources for efficient depletion of tumor-specific cancer stem cells by cytotoxic T lymphocytes (dendritic cell vaccination), adoptive transfer of chimeric antigen receptor-modified T cells, and combining immune checkpoint inhibitors with conventional therapeutic modalities. Thus, the present review captures the latest trends associated with the molecular mechanisms involved in glial tumorigenesis as well as the limitations of surgery, radiation and chemotherapy. In this article we also critically discuss the next generation molecular therapeutic strategies and their mechanisms for the successful treatment of glioma.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
