

Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a
- PMID: 28320941
- PMCID: PMC5389327
- DOI: 10.1073/pnas.1614728114
Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a
Abstract
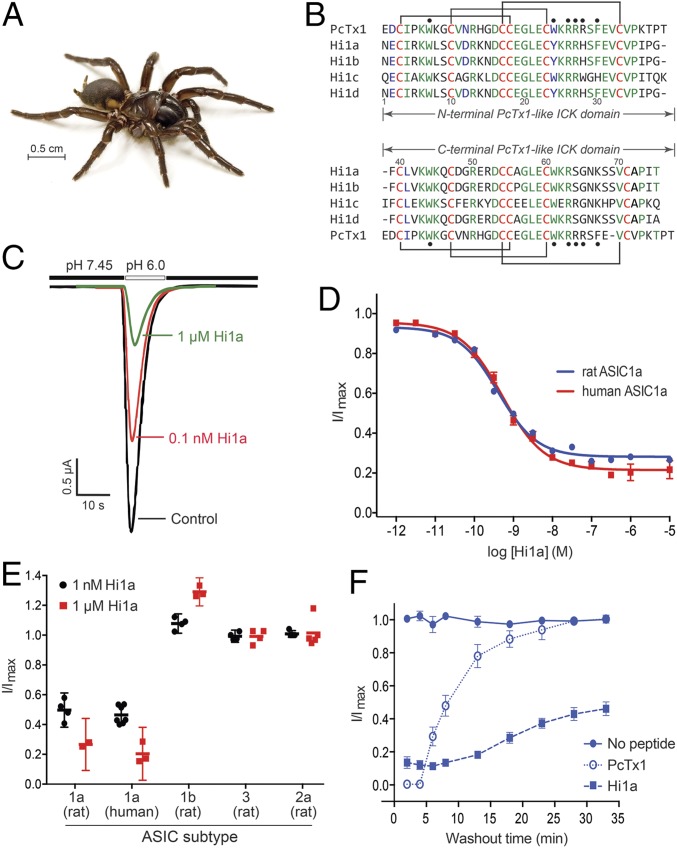
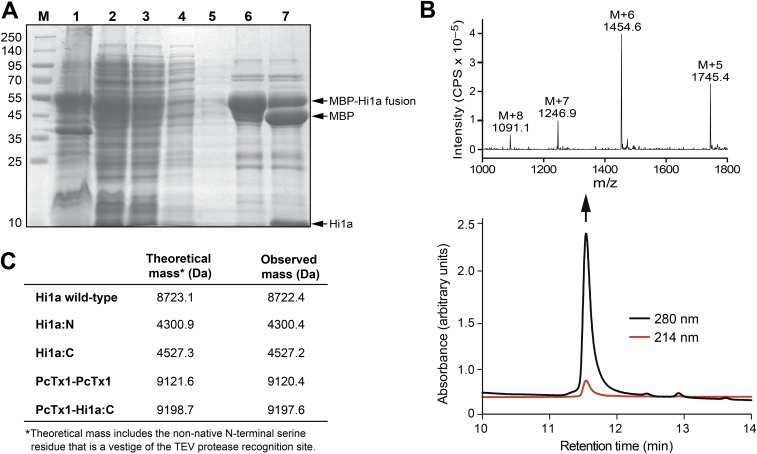
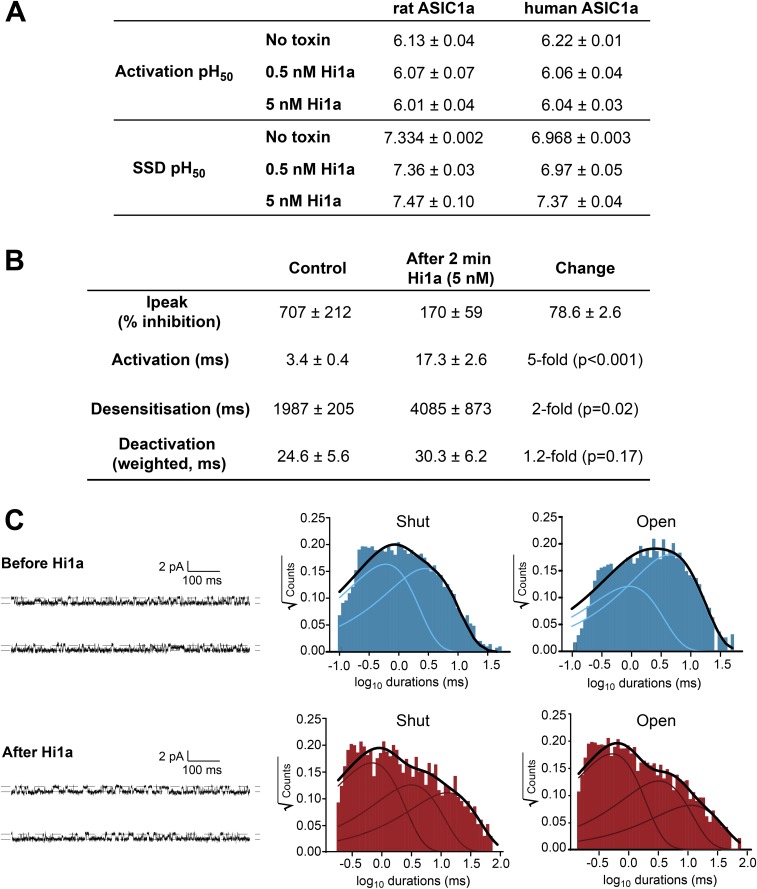
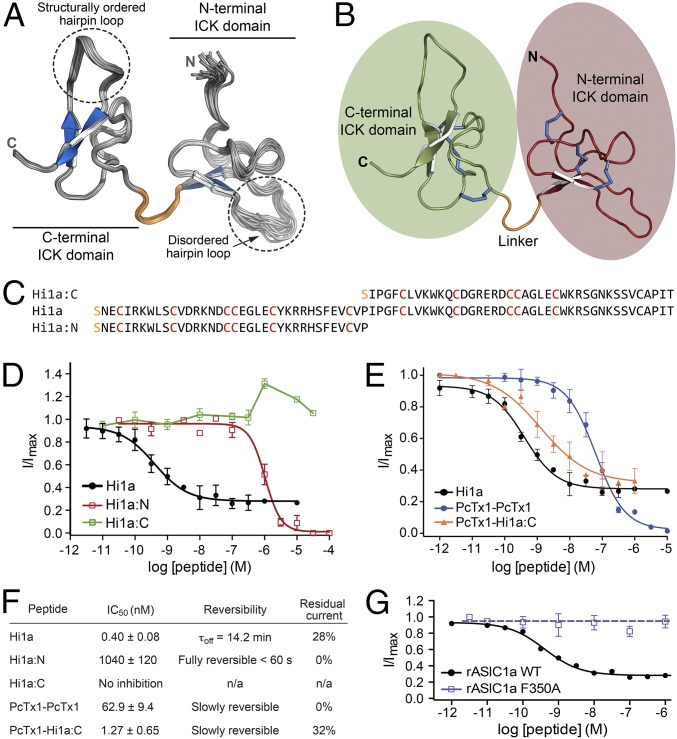
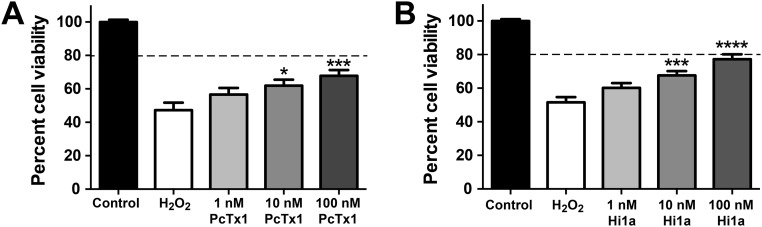
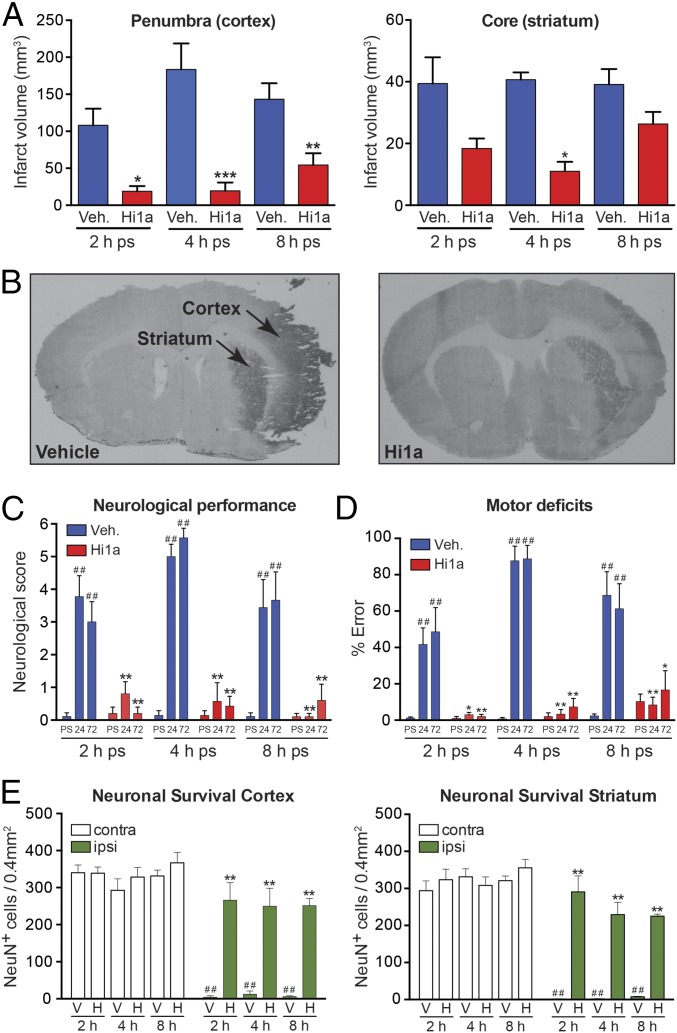
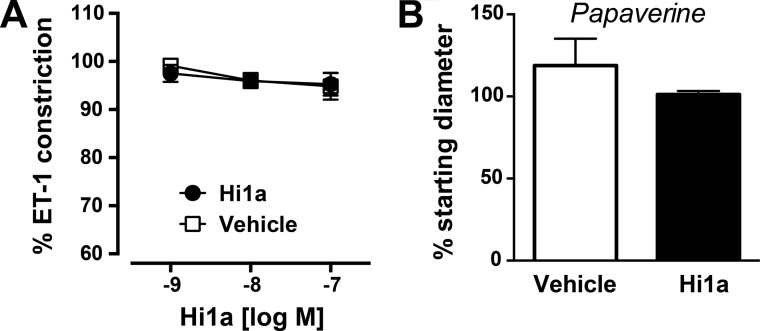
Stroke is the second-leading cause of death worldwide, yet there are no drugs available to protect the brain from stroke-induced neuronal injury. Acid-sensing ion channel 1a (ASIC1a) is the primary acid sensor in mammalian brain and a key mediator of acidosis-induced neuronal damage following cerebral ischemia. Genetic ablation and selective pharmacologic inhibition of ASIC1a reduces neuronal death following ischemic stroke in rodents. Here, we demonstrate that Hi1a, a disulfide-rich spider venom peptide, is highly neuroprotective in a focal model of ischemic stroke. Nuclear magnetic resonance structural studies reveal that Hi1a comprises two homologous inhibitor cystine knot domains separated by a short, structurally well-defined linker. In contrast with known ASIC1a inhibitors, Hi1a incompletely inhibits ASIC1a activation in a pH-independent and slowly reversible manner. Whole-cell, macropatch, and single-channel electrophysiological recordings indicate that Hi1a binds to and stabilizes the closed state of the channel, thereby impeding the transition into a conducting state. Intracerebroventricular administration to rats of a single small dose of Hi1a (2 ng/kg) up to 8 h after stroke induction by occlusion of the middle cerebral artery markedly reduced infarct size, and this correlated with improved neurological and motor function, as well as with preservation of neuronal architecture. Thus, Hi1a is a powerful pharmacological tool for probing the role of ASIC1a in acid-mediated neuronal injury and various neurological disorders, and a promising lead for the development of therapeutics to protect the brain from ischemic injury.
Keywords: acid-sensing ion channel 1a; ischemia; neuroprotection; stroke; venom peptide.
Conflict of interest statement
Conflict of interest statement: The authors' universities (The University of Queensland and Monash University) have jointly filed a patent application that covers use of the peptides described in this article (Hi1a–Hi1d).
Figures


References
-
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. - PubMed
-
- Liu R, Yuan H, Yuan F, Yang SH. Neuroprotection targeting ischemic penumbra and beyond for the treatment of ischemic stroke. Neurol Res. 2012;34(4):331–337. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
