

The Goldilocks Conundrum: NLR Inflammasome Modulation of Gastrointestinal Inflammation during Inflammatory Bowel Disease
- PMID: 28322135
- PMCID: PMC5364818
- DOI: 10.1615/CritRevImmunol.2017019158
The Goldilocks Conundrum: NLR Inflammasome Modulation of Gastrointestinal Inflammation during Inflammatory Bowel Disease
Abstract
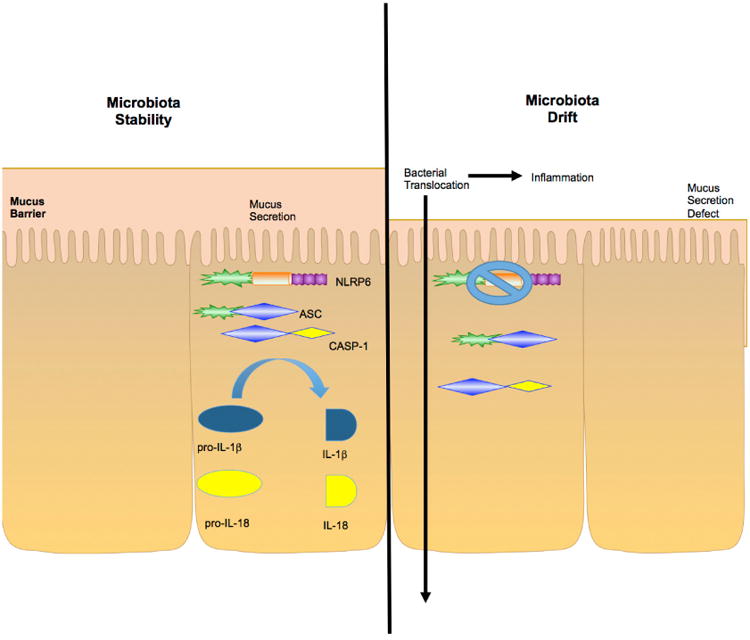
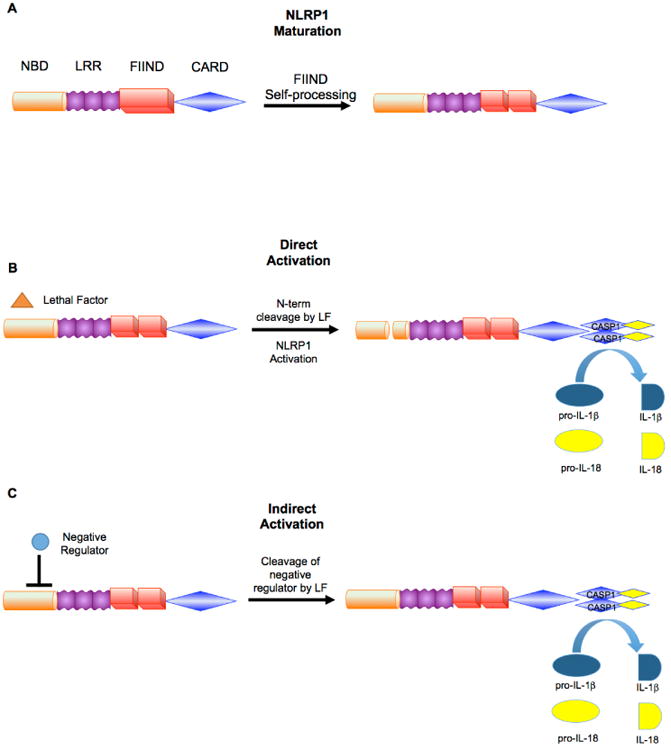
Recent advances have revealed significant insight into inflammatory bowel disease (IBD) pathobiology. Ulcerative colitis and Crohn's disease, the chronic relapsing clinical manifestations of IBD, are complex disorders with genetic and environmental influences. These diseases are associated with the dysregulation of immune tolerance, excessive inflammation, and damage to the epithelial cell barrier. Increasing evidence indicates that pattern recognition receptors, including Toll-like receptors (TLRs) and nucleotide-binding domain and leucine-rich repeat-containing proteins (NLRs), function to maintain immune system homeostasis, modulate the gastrointestinal microbiome, and promote proper intestinal epithelial cell regeneration and repair. New insights have revealed that NLR family members are essential components in maintaining this immune system homeostasis. To date, the vast majority of studies associated with NLRs have focused on family members that form a multiprotein signaling platform called the inflammasome. These signaling complexes are responsible for the cleavage and activation of the potent pleotropic cytokines IL-1β and IL-18, and they facilitate a unique form of cell death defined as pyroptosis. In this review, we summarize the current paradigms associated with NLR inflammasome maintenance of immune system homeostasis in the gastrointestinal system. New concepts related to canonical and noncanonical inflammasome signaling, as well as the implications of classical and alternative inflammasomes in IBD pathogenesis, are also reviewed.
Figures






Similar articles
-
[Inflammatory bowel diseases and inflammasome].Korean J Gastroenterol. 2011 Dec;58(6):300-10. doi: 10.4166/kjg.2011.58.6.300. Korean J Gastroenterol. 2011. PMID: 22198227 Review. Korean.
-
NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways.Arch Biochem Biophys. 2019 Jul 30;670:4-14. doi: 10.1016/j.abb.2019.02.008. Epub 2019 Feb 14. Arch Biochem Biophys. 2019. PMID: 30772258 Review.
-
NLRP3 Inflammasome and Inflammatory Bowel Disease.Front Immunol. 2019 Feb 28;10:276. doi: 10.3389/fimmu.2019.00276. eCollection 2019. Front Immunol. 2019. PMID: 30873162 Free PMC article. Review.
-
Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health.Int J Mol Sci. 2017 Nov 9;18(11):2379. doi: 10.3390/ijms18112379. Int J Mol Sci. 2017. PMID: 29120406 Free PMC article. Review.
-
The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis.J Immunol. 2015 Apr 1;194(7):3369-80. doi: 10.4049/jimmunol.1402098. Epub 2015 Feb 27. J Immunol. 2015. PMID: 25725098 Free PMC article.
Cited by
-
GDF-11 Protects the Traumatically Injured Spinal Cord by Suppressing Pyroptosis and Necroptosis via TFE3-Mediated Autophagy Augmentation.Oxid Med Cell Longev. 2021 Oct 19;2021:8186877. doi: 10.1155/2021/8186877. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34712387 Free PMC article.
-
An integrative network-based approach to identify novel disease genes and pathways: a case study in the context of inflammatory bowel disease.BMC Bioinformatics. 2018 Jul 13;19(1):264. doi: 10.1186/s12859-018-2251-x. BMC Bioinformatics. 2018. PMID: 30005591 Free PMC article.
-
Interleukin-18: Biological properties and role in disease pathogenesis.Immunol Rev. 2018 Jan;281(1):138-153. doi: 10.1111/imr.12616. Immunol Rev. 2018. PMID: 29247988 Free PMC article. Review.
-
Relationship between neutrophil-lymphocyte ratio and short-term prognosis in the chronic obstructive pulmonary patients with acute exacerbation.Biosci Rep. 2019 May 14;39(5):BSR20190675. doi: 10.1042/BSR20190675. Print 2019 May 31. Biosci Rep. 2019. PMID: 31015366 Free PMC article.
-
The role of microbiota in the development of colorectal cancer.Int J Cancer. 2019 Oct 15;145(8):2032-2041. doi: 10.1002/ijc.32017. Epub 2019 Jan 15. Int J Cancer. 2019. PMID: 30474116 Free PMC article. Review.
References
-
- Walsh D, McCarthy J, O'Driscoll C, Melgar S. Pattern recognition receptors—molecular orchestrators of Inflammation in Inflammatory bowel disease. Cytokine Growth Factor Rev. 2013;24(2):91–104. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous