

doi: 10.1371/journal.pone.0174250.
eCollection 2017.
IsoSel: Protein Isoform Selector for phylogenetic reconstructions
Affiliations
- PMID: 28323858
- PMCID: PMC5360266
- DOI: 10.1371/journal.pone.0174250
Item in Clipboard
IsoSel: Protein Isoform Selector for phylogenetic reconstructions
PLoS One.
.
Abstract
The reliability of molecular phylogenies is strongly dependent on the quality of the assembled datasets. In the case of eukaryotes, the selection of only one protein isoform per genomic locus is mandatory to avoid biases linked to redundancy. Here, we present IsoSel, a tool devoted to the selection of alternative isoforms in the context of phylogenetic reconstruction. It provides a better alternative to the widely used approach consisting in the selection of the longest isoforms and it performs better than Guidance, its only available counterpart. IsoSel is publicly available at http://doua.prabi.fr/software/isosel.
Conflict of interest statement
Figures
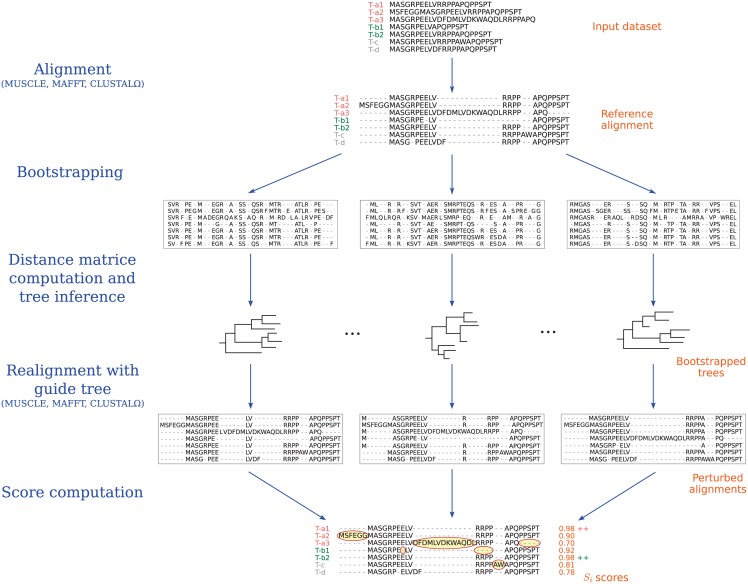
Schematic representation of the different steps performed during an IsoSel run. T-x represent alternatives isoforms generated by a same gene x. In this example, isoforms a1 and b2 are selected for the genes a and b, respectively.
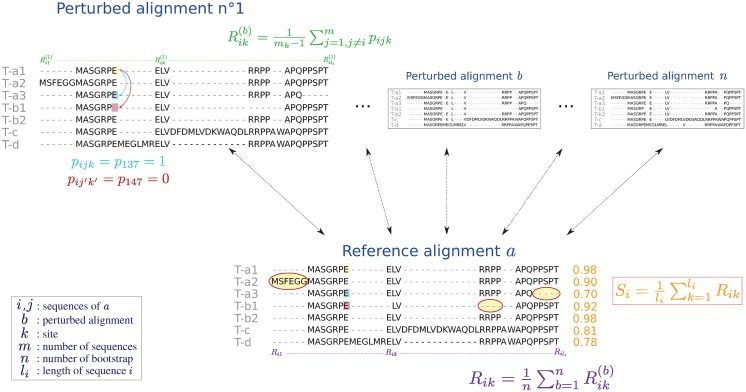
Example of score computation for four genes (a, b, c and d) producing three (a), two (b) and no (c and d) alternative isoforms.
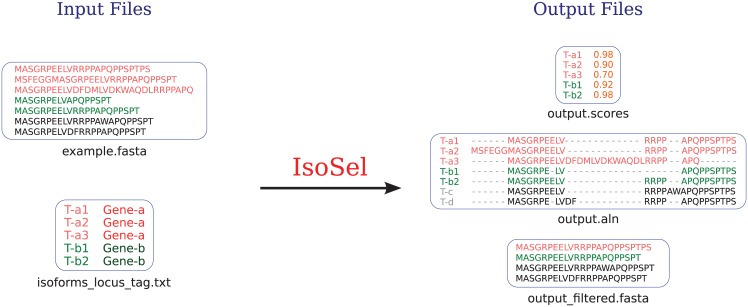
IsoSel minimal input requirement is an unaligned protein sequence dataset in Fasta format (example.fasta ). The two output files generated contain the alignment (output.aln ) and the sequences scores (output.scores or output.DistanceScore if the -DS option is used). Optionally, the user can provide a file containing the genomic origin of the input sequences (isoforms_locus_tag.txt ). In this case, an additional file containing, for each locus, the sequence having the highest score is created (output_filtered.fasta ).
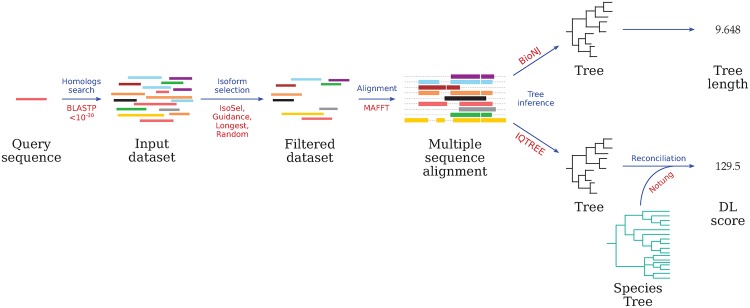
For a given human protein from UniProtKB, a BLASTP search is performed. The alternative isoforms detected for each set of homologs are then selected using either the longest isoform, a random choice, Guidance or IsoSel. Then the sets are aligned and the corresponding gene trees are inferred by BioNJ and IQ-TREE for computing tree lengths and DL scores, respectively. For each step, algorithms used are indicated in red.
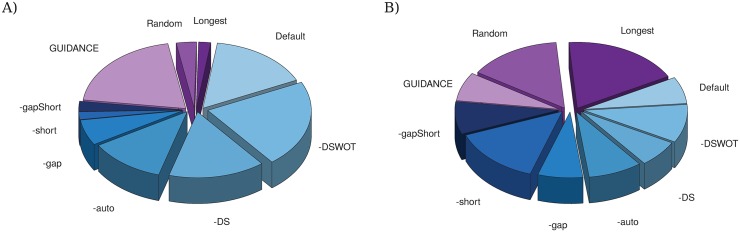
Charts are proportional to the number of: A) the shortest trees; and B) the trees with the lower DL score obtained with the different options and programs. Charts in shades of blue correspond to the different IsoSel options.
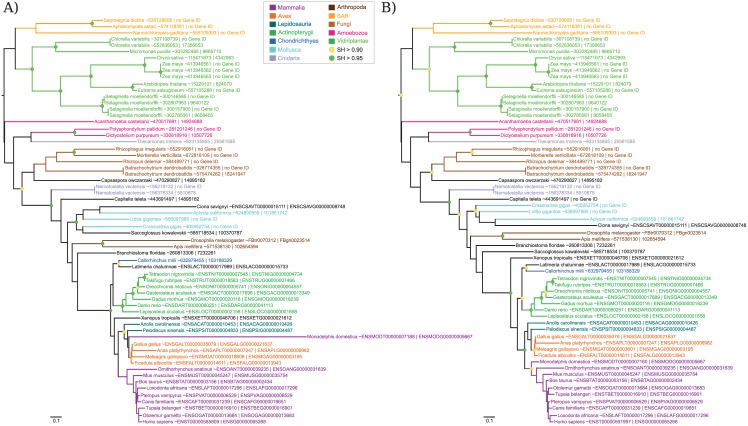
Isoform selection was done by selecting the longest isoform (A) and by running IsoSel with its default parameters (B). Sequences are colored according to their taxonomic classification. Green and yellow circles correspond to nodes with SH > 0.95 and SH > 0.90, respectively. The scale bar represents the average number of substitutions per site.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
