

Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation
- PMID: 28325822
- PMCID: PMC5648539
- DOI: 10.1126/scisignal.aah5756
Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation
Abstract
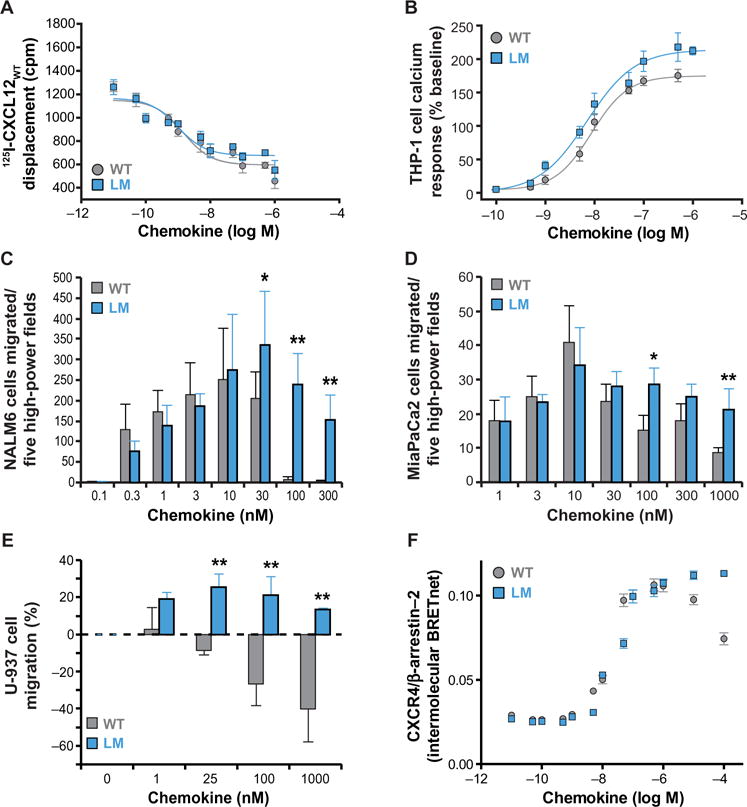
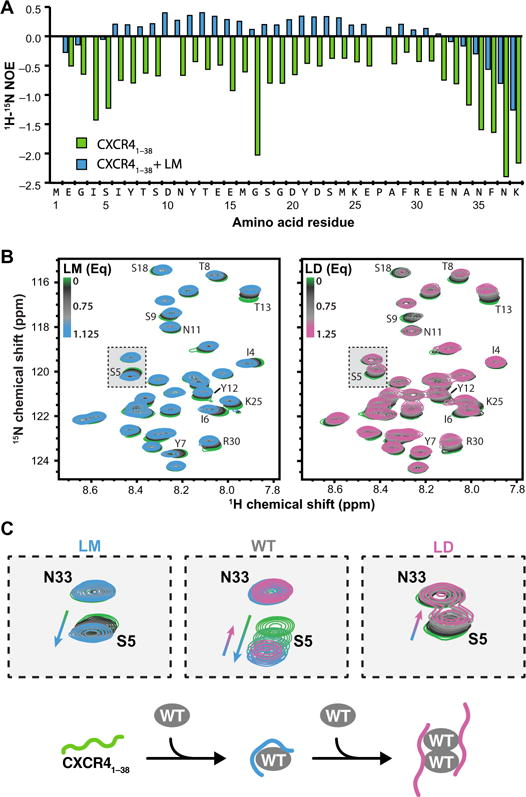
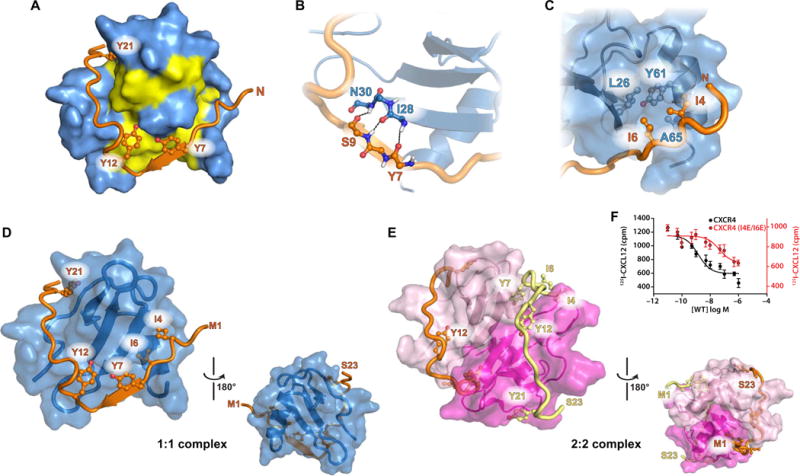
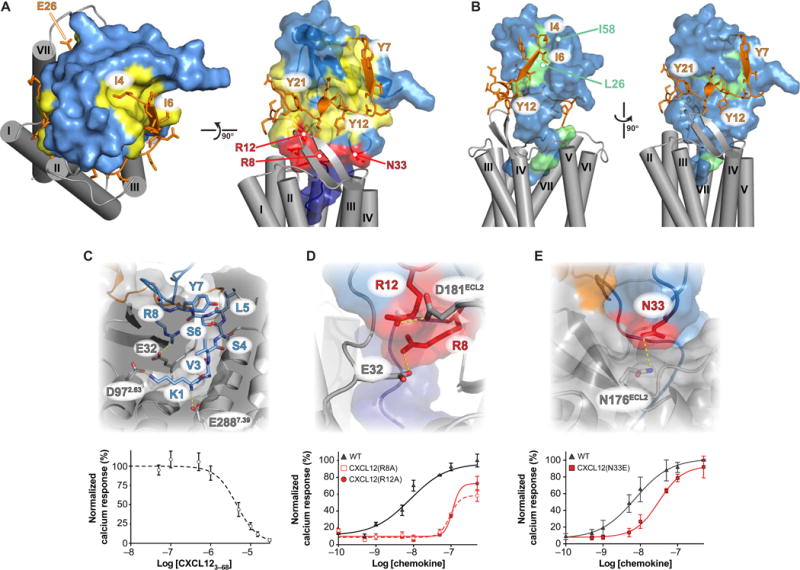
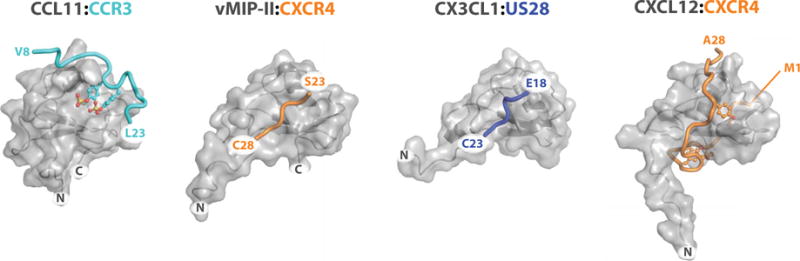
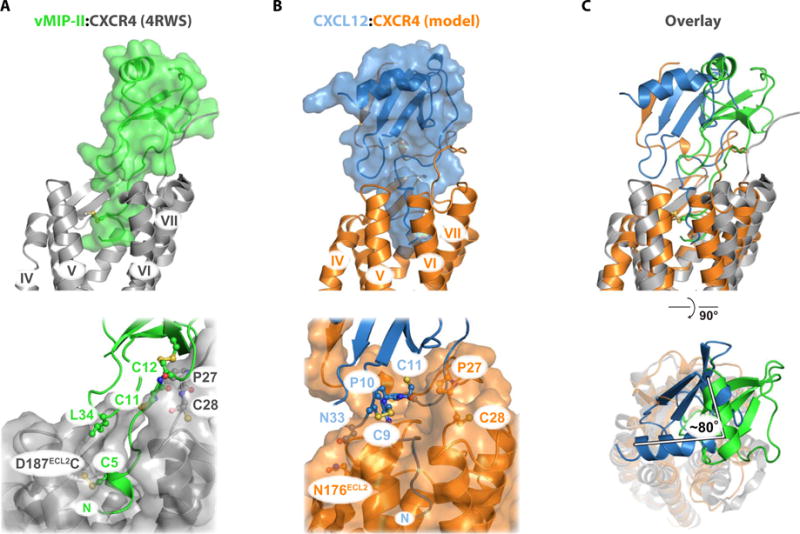
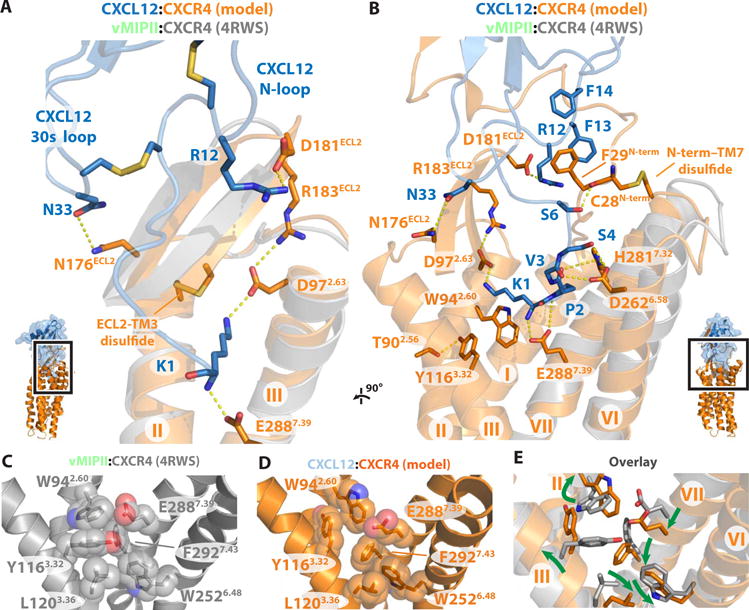
Chemokines orchestrate cell migration for development, immune surveillance, and disease by binding to cell surface heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs). The array of interactions between the nearly 50 chemokines and their 20 GPCR targets generates an extensive signaling network to which promiscuity and biased agonism add further complexity. The receptor CXCR4 recognizes both monomeric and dimeric forms of the chemokine CXCL12, which is a distinct example of ligand bias in the chemokine family. We demonstrated that a constitutively monomeric CXCL12 variant reproduced the G protein-dependent and β-arrestin-dependent responses that are associated with normal CXCR4 signaling and lead to cell migration. In addition, monomeric CXCL12 made specific contacts with CXCR4 that are not present in the structure of the receptor in complex with a dimeric form of CXCL12, a biased agonist that stimulates only G protein-dependent signaling. We produced an experimentally validated model of an agonist-bound chemokine receptor that merged a nuclear magnetic resonance-based structure of monomeric CXCL12 bound to the amino terminus of CXCR4 with a crystal structure of the transmembrane domains of CXCR4. The large CXCL12:CXCR4 protein-protein interface revealed by this structure identified previously uncharacterized functional interactions that fall outside of the classical "two-site model" for chemokine-receptor recognition. Our model suggests a mechanistic hypothesis for how interactions on the extracellular face of the receptor may stimulate the conformational changes required for chemokine receptor-mediated signal transduction.
Copyright © 2017, American Association for the Advancement of Science.
Conflict of interest statement
Figures
References
-
- Ohnishi Y, Senda T, Nandhagopal N, Sugimoto K, Shioda T, Nagal Y, Mitsui Y. Crystal structure of recombinant native SDF-1α with additional mutagenesis studies: An attempt at a more comprehensive interpretation of accumulated structure-activity relationship data. J Interferon Cytokine Res. 2000;20:691–700. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
