

Lessons learned from additional research analyses of unsolved clinical exome cases
- PMID: 28327206
- PMCID: PMC5361813
- DOI: 10.1186/s13073-017-0412-6
Lessons learned from additional research analyses of unsolved clinical exome cases
Abstract
Background: Given the rarity of most single-gene Mendelian disorders, concerted efforts of data exchange between clinical and scientific communities are critical to optimize molecular diagnosis and novel disease gene discovery.
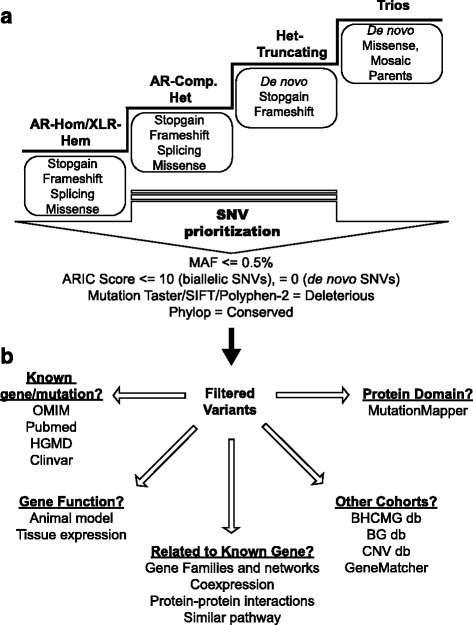
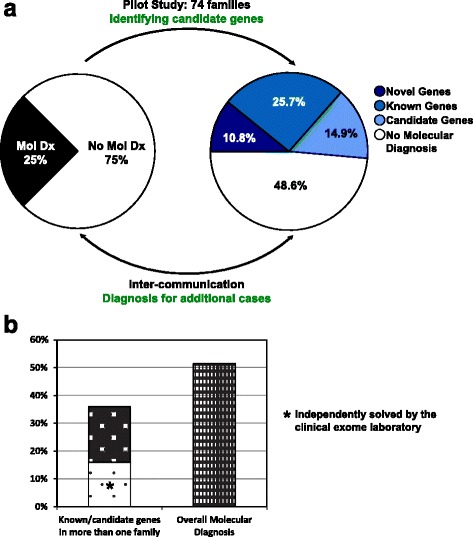
Methods: We designed and implemented protocols for the study of cases for which a plausible molecular diagnosis was not achieved in a clinical genomics diagnostic laboratory (i.e. unsolved clinical exomes). Such cases were recruited to a research laboratory for further analyses, in order to potentially: (1) accelerate novel disease gene discovery; (2) increase the molecular diagnostic yield of whole exome sequencing (WES); and (3) gain insight into the genetic mechanisms of disease. Pilot project data included 74 families, consisting mostly of parent-offspring trios. Analyses performed on a research basis employed both WES from additional family members and complementary bioinformatics approaches and protocols.
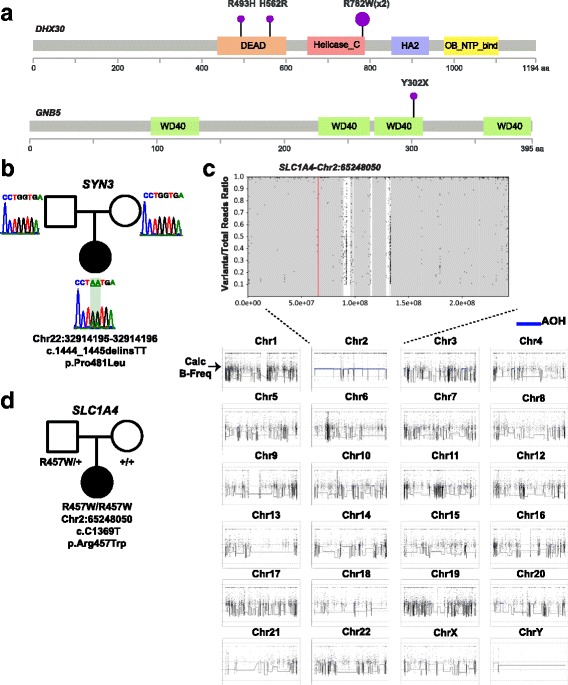
Results: Analysis of all possible modes of Mendelian inheritance, focusing on both single nucleotide variants (SNV) and copy number variant (CNV) alleles, yielded a likely contributory variant in 36% (27/74) of cases. If one includes candidate genes with variants identified within a single family, a potential contributory variant was identified in a total of ~51% (38/74) of cases enrolled in this pilot study. The molecular diagnosis was achieved in 30/63 trios (47.6%). Besides this, the analysis workflow yielded evidence for pathogenic variants in disease-associated genes in 4/6 singleton cases (66.6%), 1/1 multiplex family involving three affected siblings, and 3/4 (75%) quartet families. Both the analytical pipeline and the collaborative efforts between the diagnostic and research laboratories provided insights that allowed recent disease gene discoveries (PURA, TANGO2, EMC1, GNB5, ATAD3A, and MIPEP) and increased the number of novel genes, defined in this study as genes identified in more than one family (DHX30 and EBF3).
Conclusion: An efficient genomics pipeline in which clinical sequencing in a diagnostic laboratory is followed by the detailed reanalysis of unsolved cases in a research environment, supplemented with WES data from additional family members, and subject to adjuvant bioinformatics analyses including relaxed variant filtering parameters in informatics pipelines, can enhance the molecular diagnostic yield and provide mechanistic insights into Mendelian disorders. Implementing these approaches requires collaborative clinical molecular diagnostic and research efforts.
Figures
References
-
- Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–62. doi: 10.1097/GIM.0b013e3182088158. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
