

Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders
- PMID: 28327575
- PMCID: PMC5477361
- DOI: 10.1038/ejhg.2017.32
Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders
Abstract
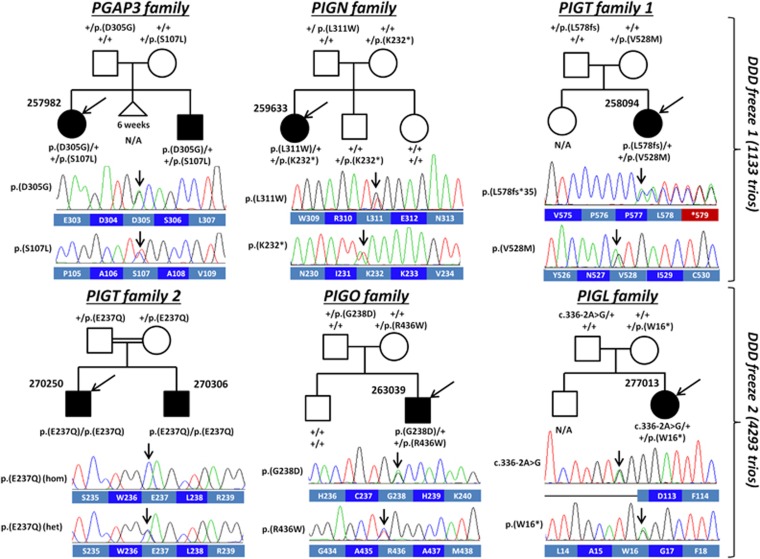
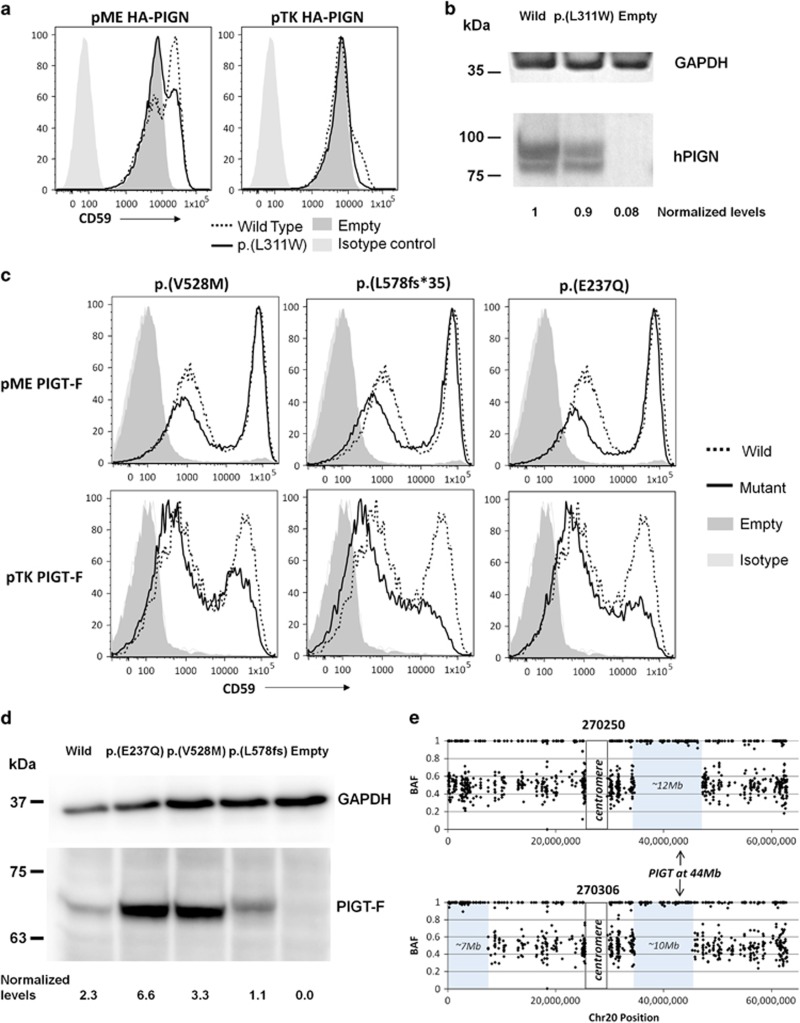
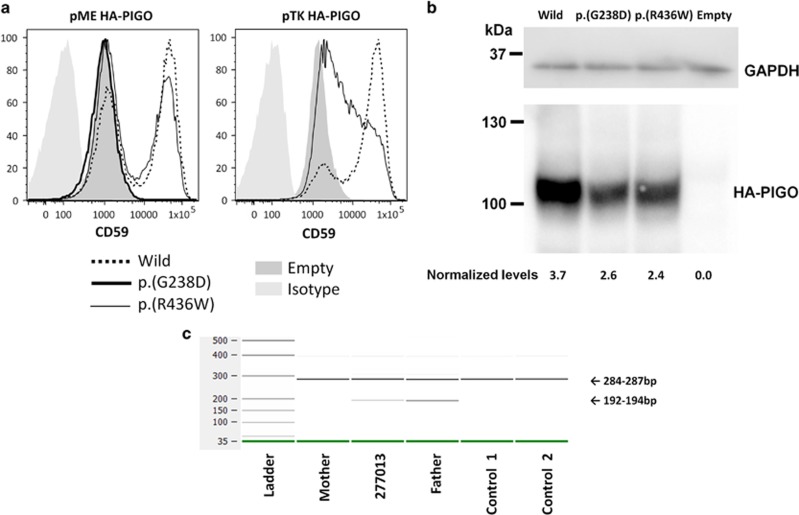
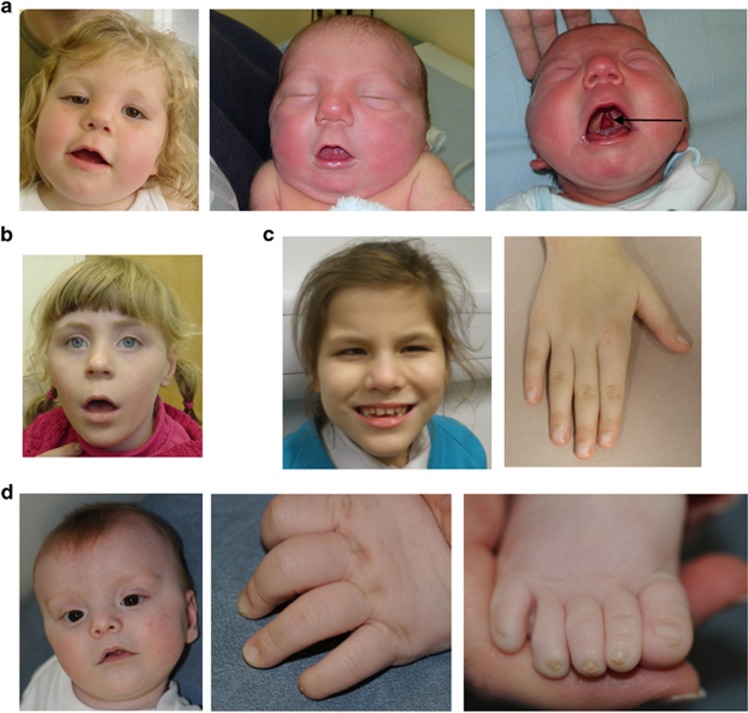
Over 150 different proteins attach to the plasma membrane using glycosylphosphatidylinositol (GPI) anchors. Mutations in 18 genes that encode components of GPI-anchor biogenesis result in a phenotypic spectrum that includes learning disability, epilepsy, microcephaly, congenital malformations and mild dysmorphic features. To determine the incidence of GPI-anchor defects, we analysed the exome data from 4293 parent-child trios recruited to the Deciphering Developmental Disorders (DDD) study. All probands recruited had a neurodevelopmental disorder. We searched for variants in 31 genes linked to GPI-anchor biogenesis and detected rare biallelic variants in PGAP3, PIGN, PIGT (n=2), PIGO and PIGL, providing a likely diagnosis for six families. In five families, the variants were in a compound heterozygous configuration while in a consanguineous Afghani kindred, a homozygous c.709G>C; p.(E237Q) variant in PIGT was identified within 10-12 Mb of autozygosity. Validation and segregation analysis was performed using Sanger sequencing. Across the six families, five siblings were available for testing and in all cases variants co-segregated consistent with them being causative. In four families, abnormal alkaline phosphatase results were observed in the direction expected. FACS analysis of knockout HEK293 cells that had been transfected with wild-type or mutant cDNA constructs demonstrated that the variants in PIGN, PIGT and PIGO all led to reduced activity. Splicing assays, performed using leucocyte RNA, showed that a c.336-2A>G variant in PIGL resulted in exon skipping and p.D113fs*2. Our results strengthen recently reported disease associations, suggest that defective GPI-anchor biogenesis may explain ~0.15% of individuals with developmental disorders and highlight the benefits of data sharing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Takeda J, Miyata T, Kawagoe K et al: Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 1993; 73: 703–711. - PubMed
-
- Belet S, Fieremans N, Yuan X et al: Early frameshift mutation in PIGA identified in a large XLID family without neonatal lethality. Hum Mutat 2014; 35: 350–355. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
