

Endogenous opioids regulate moment-to-moment neuronal communication and excitability
- PMID: 28327612
- PMCID: PMC5364458
- DOI: 10.1038/ncomms14611
Endogenous opioids regulate moment-to-moment neuronal communication and excitability
Abstract
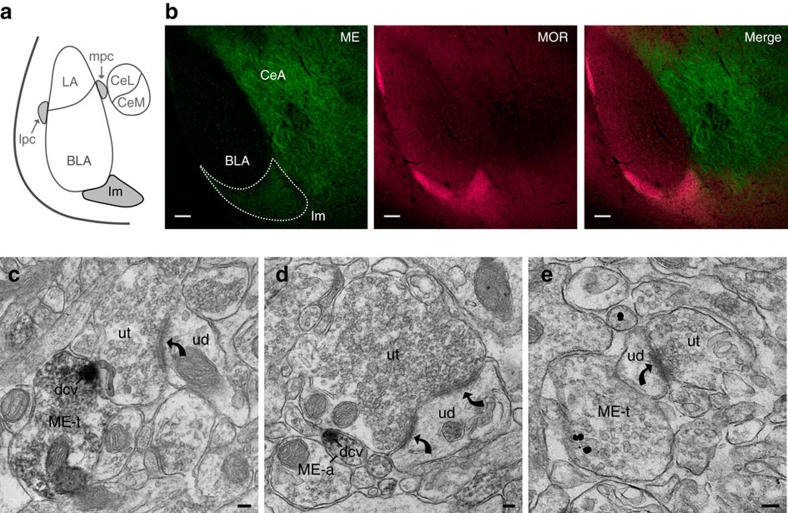
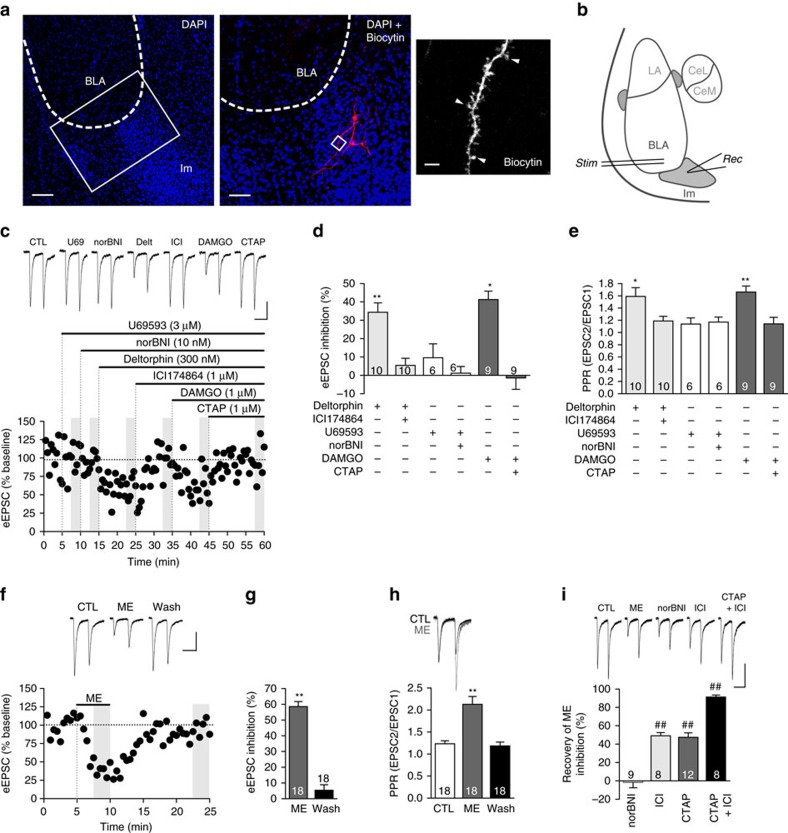
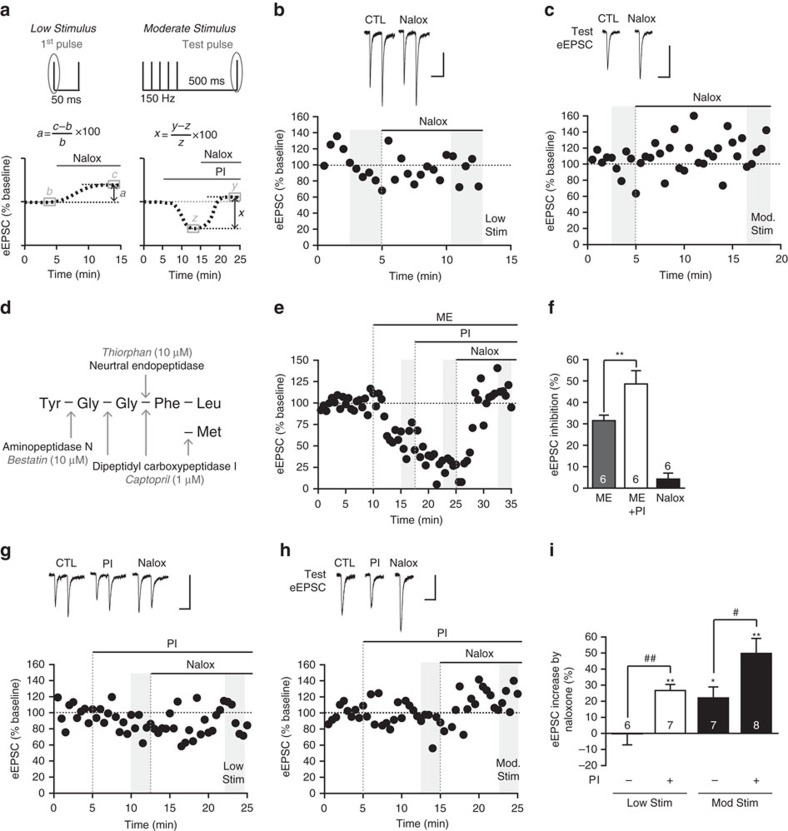
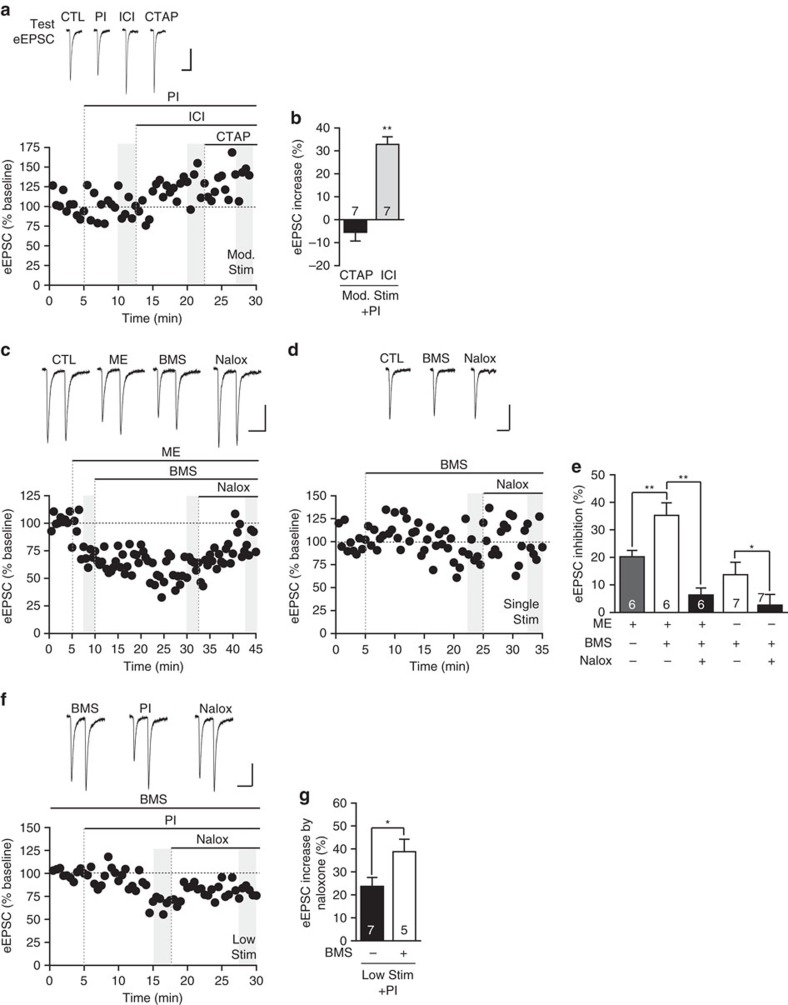
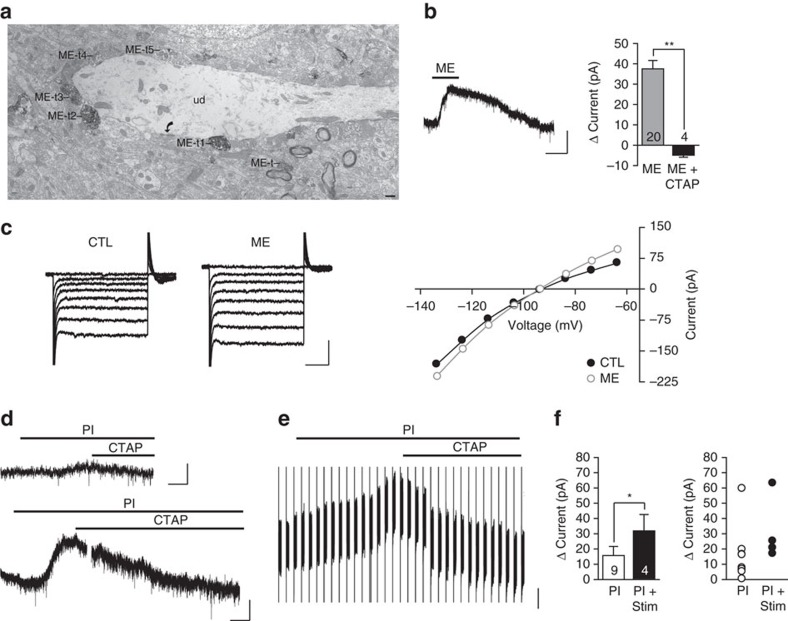
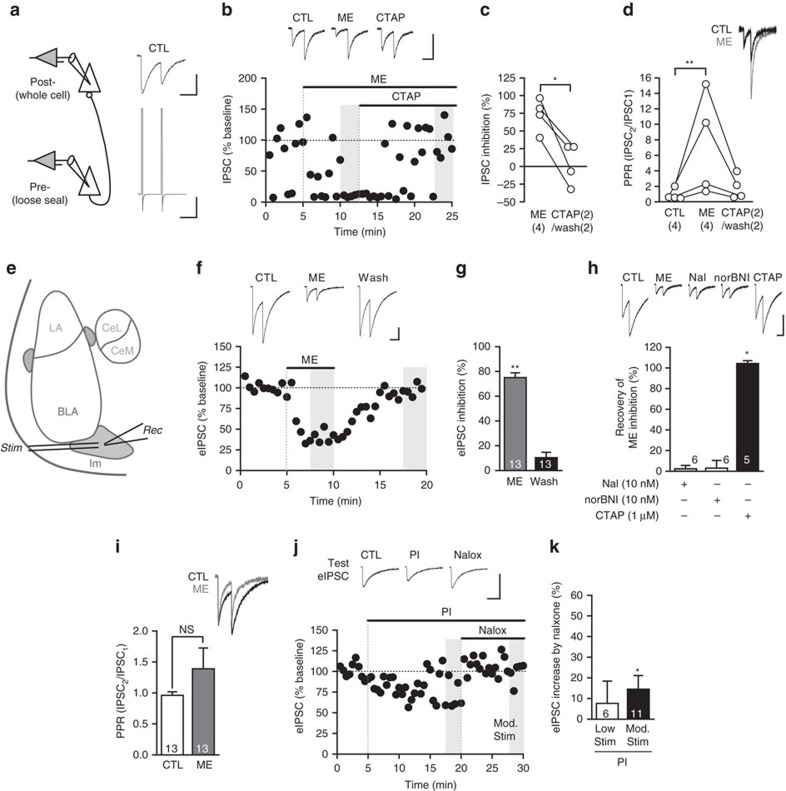
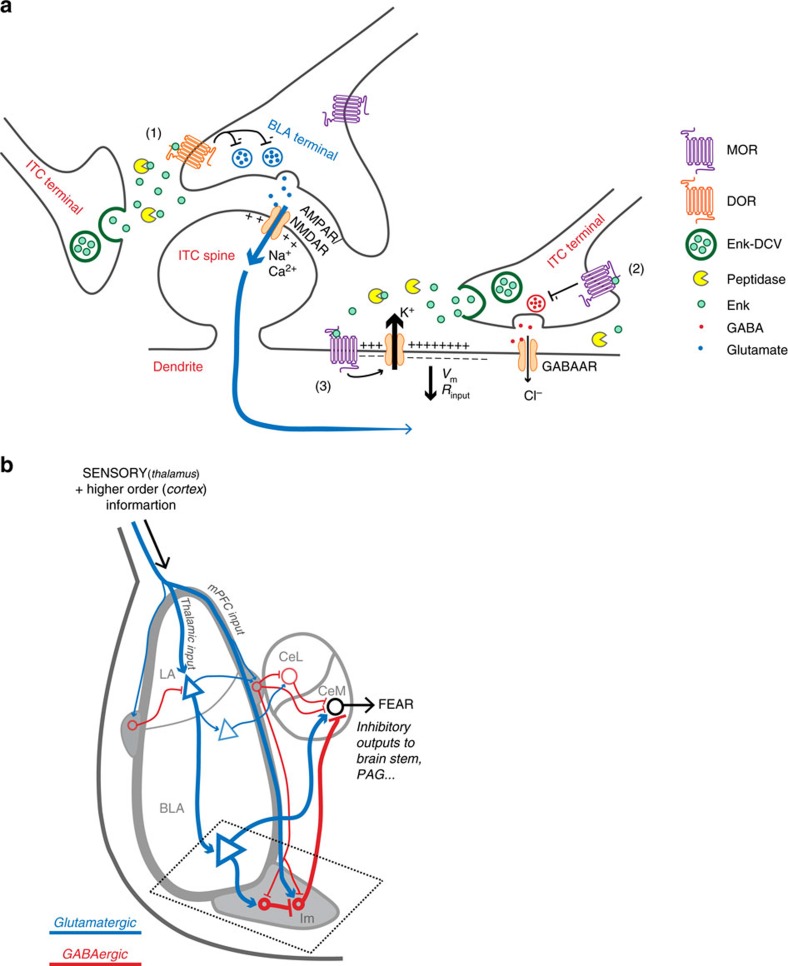
Fear and emotional learning are modulated by endogenous opioids but the cellular basis for this is unknown. The intercalated cells (ITCs) gate amygdala output and thus regulate the fear response. Here we find endogenous opioids are released by synaptic stimulation to act via two distinct mechanisms within the main ITC cluster. Endogenously released opioids inhibit glutamate release through the δ-opioid receptor (DOR), an effect potentiated by a DOR-positive allosteric modulator. Postsynaptically, the opioids activate a potassium conductance through the μ-opioid receptor (MOR), suggesting for the first time that endogenously released opioids directly regulate neuronal excitability. Ultrastructural localization of endogenous ligands support these functional findings. This study demonstrates a new role for endogenously released opioids as neuromodulators engaged by synaptic activity to regulate moment-to-moment neuronal communication and excitability. These distinct actions through MOR and DOR may underlie the opposing effect of these receptor systems on anxiety and fear.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Phelps E. A. & LeDoux J. E. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48, 175–187 (2005). - PubMed
-
- Konig M. et al.. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature 383, 535–538 (1996). - PubMed
-
- Nieto M. M., Guen S. L., Kieffer B. L., Roques B. P. & Noble F. Physiological control of emotion-related behaviors by endogenous enkephalins involves essentially the delta opioid receptors. Neuroscience 135, 305–313 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
