

MinION™ nanopore sequencing of environmental metagenomes: a synthetic approach
- PMID: 28327976
- PMCID: PMC5467020
- DOI: 10.1093/gigascience/gix007
MinION™ nanopore sequencing of environmental metagenomes: a synthetic approach
Abstract
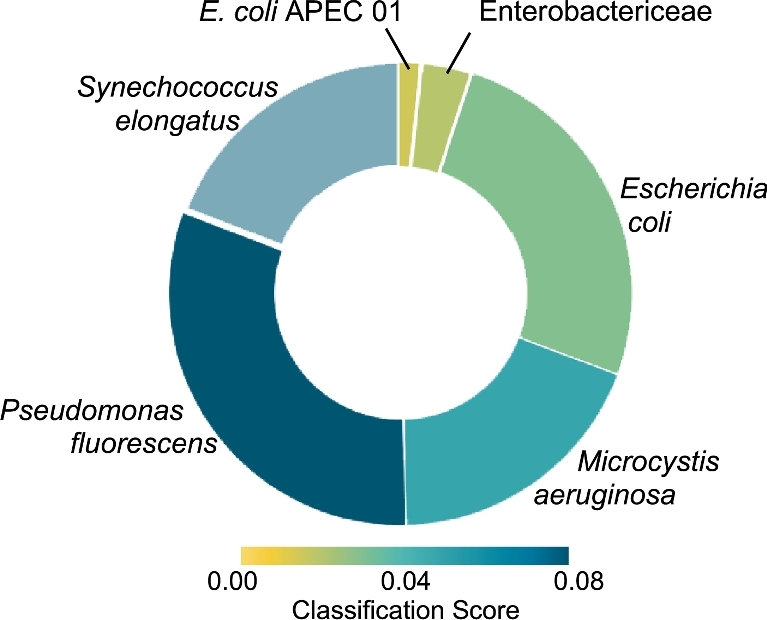
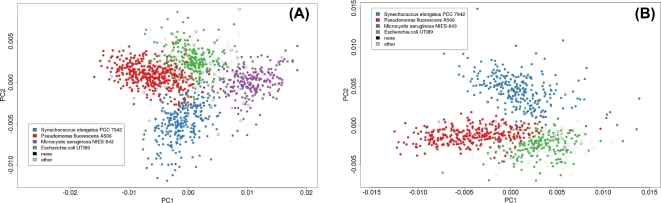
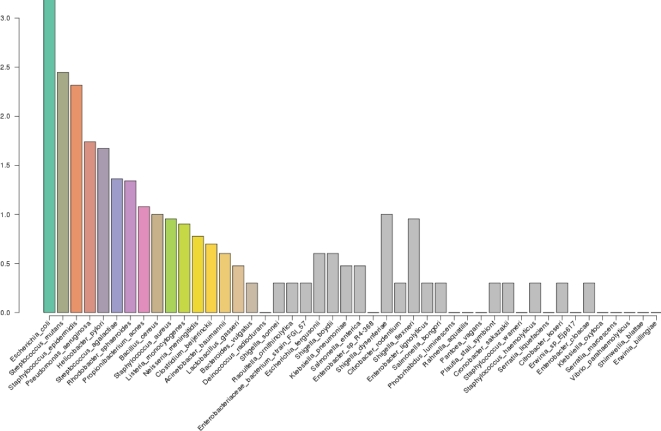
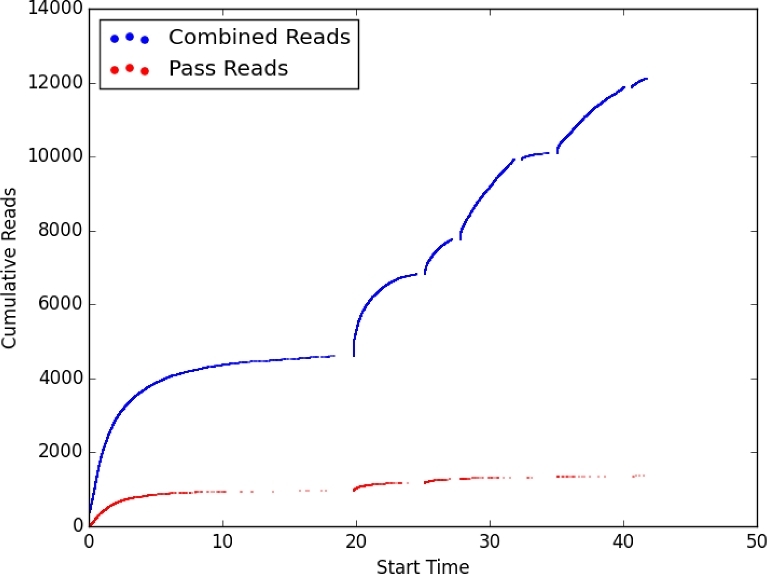
Environmental metagenomic analysis is typically accomplished by assigning taxonomy and/or function from whole genome sequencing or 16S amplicon sequences. Both of these approaches are limited, however, by read length, among other technical and biological factors. A nanopore-based sequencing platform, MinION™, produces reads that are ≥1 × 104 bp in length, potentially providing for more precise assignment, thereby alleviating some of the limitations inherent in determining metagenome composition from short reads. We tested the ability of sequence data produced by MinION (R7.3 flow cells) to correctly assign taxonomy in single bacterial species runs and in three types of low-complexity synthetic communities: a mixture of DNA using equal mass from four species, a community with one relatively rare (1%) and three abundant (33% each) components, and a mixture of genomic DNA from 20 bacterial strains of staggered representation. Taxonomic composition of the low-complexity communities was assessed by analyzing the MinION sequence data with three different bioinformatic approaches: Kraken, MG-RAST, and One Codex. Results: Long read sequences generated from libraries prepared from single strains using the version 5 kit and chemistry, run on the original MinION device, yielded as few as 224 to as many as 3497 bidirectional high-quality (2D) reads with an average overall study length of 6000 bp. For the single-strain analyses, assignment of reads to the correct genus by different methods ranged from 53.1% to 99.5%, assignment to the correct species ranged from 23.9% to 99.5%, and the majority of misassigned reads were to closely related organisms. A synthetic metagenome sequenced with the same setup yielded 714 high quality 2D reads of approximately 5500 bp that were up to 98% correctly assigned to the species level. Synthetic metagenome MinION libraries generated using version 6 kit and chemistry yielded from 899 to 3497 2D reads with lengths averaging 5700 bp with up to 98% assignment accuracy at the species level. The observed community proportions for “equal” and “rare” synthetic libraries were close to the known proportions, deviating from 0.1% to 10% across all tests. For a 20-species mock community with staggered contributions, a sequencing run detected all but 3 species (each included at <0.05% of DNA in the total mixture), 91% of reads were assigned to the correct species, 93% of reads were assigned to the correct genus, and >99% of reads were assigned to the correct family. Conclusions: At the current level of output and sequence quality (just under 4 × 103 2D reads for a synthetic metagenome), MinION sequencing followed by Kraken or One Codex analysis has the potential to provide rapid and accurate metagenomic analysis where the consortium is comprised of a limited number of taxa. Important considerations noted in this study included: high sensitivity of the MinION platform to the quality of input DNA, high variability of sequencing results across libraries and flow cells, and relatively small numbers of 2D reads per analysis limit. Together, these limited detection of very rare components of the microbial consortia, and would likely limit the utility of MinION for the sequencing of high-complexity metagenomic communities where thousands of taxa are expected. Furthermore, the limitations of the currently available data analysis tools suggest there is considerable room for improvement in the analytical approaches for the characterization of microbial communities using long reads. Nevertheless, the fact that the accurate taxonomic assignment of high-quality reads generated by MinION is approaching 99.5% and, in most cases, the inferred community structure mirrors the known proportions of a synthetic mixture warrants further exploration of practical application to environmental metagenomics as the platform continues to develop and improve. With further improvement in sequence throughput and error rate reduction, this platform shows great promise for precise real-time analysis of the composition and structure of more complex microbial communities.
Keywords: Long-read sequencing; Metagenome; MinION™; Oxford Nanopore Technologies; Whole-genome sequencing.
© The Author 2017. Published by Oxford University Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
