

The blood DNA virome in 8,000 humans
- PMID: 28328962
- PMCID: PMC5378407
- DOI: 10.1371/journal.ppat.1006292
The blood DNA virome in 8,000 humans
Abstract
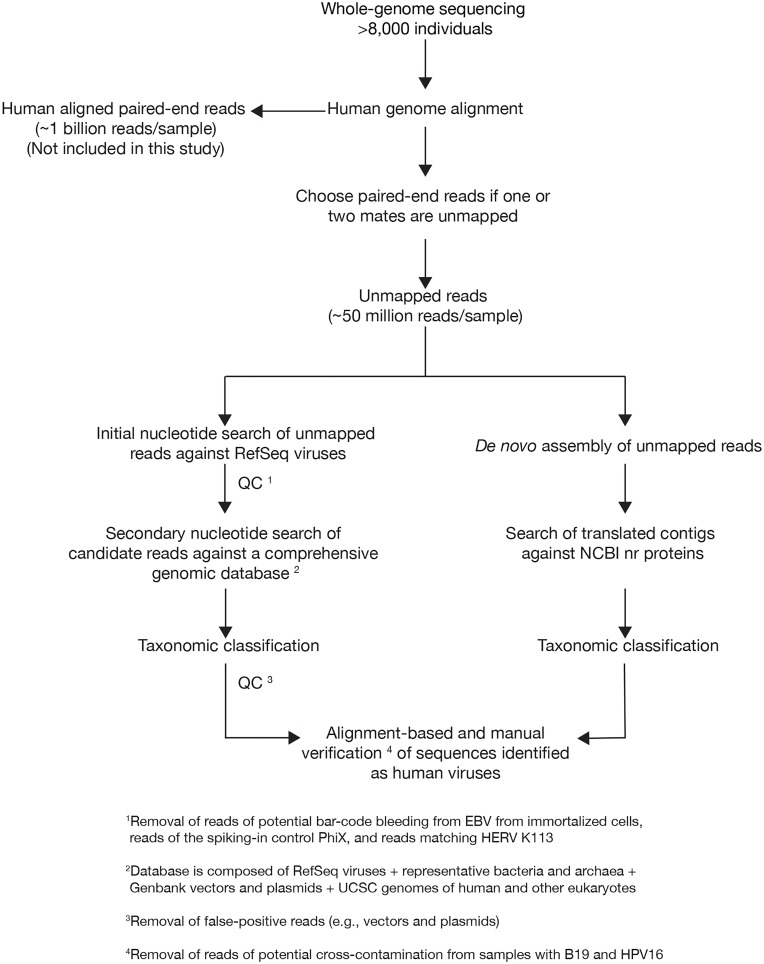
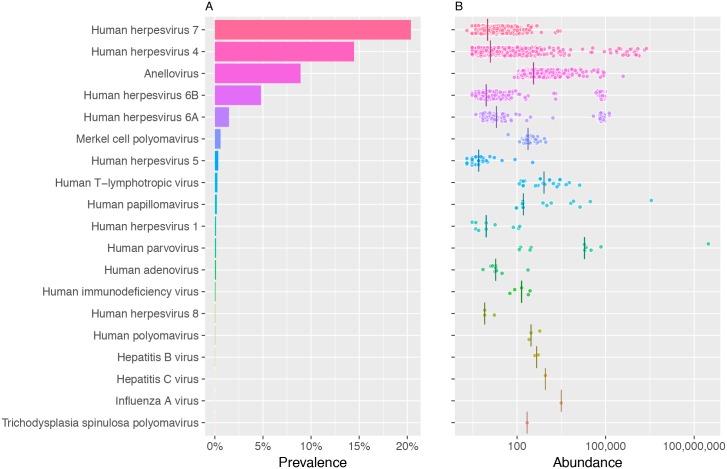
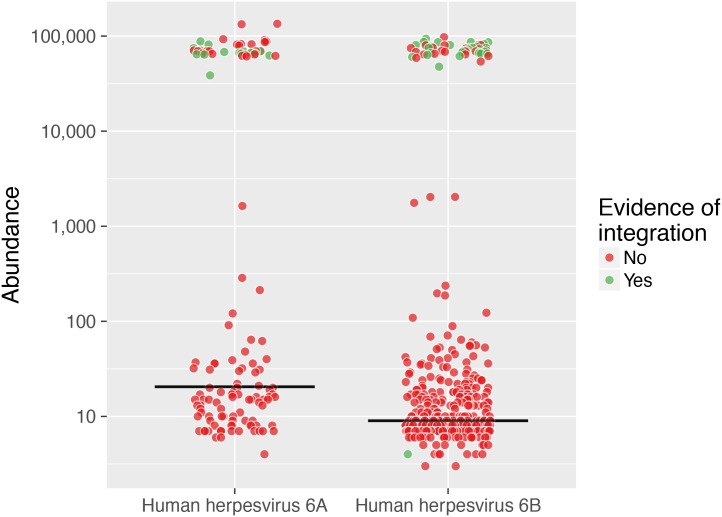
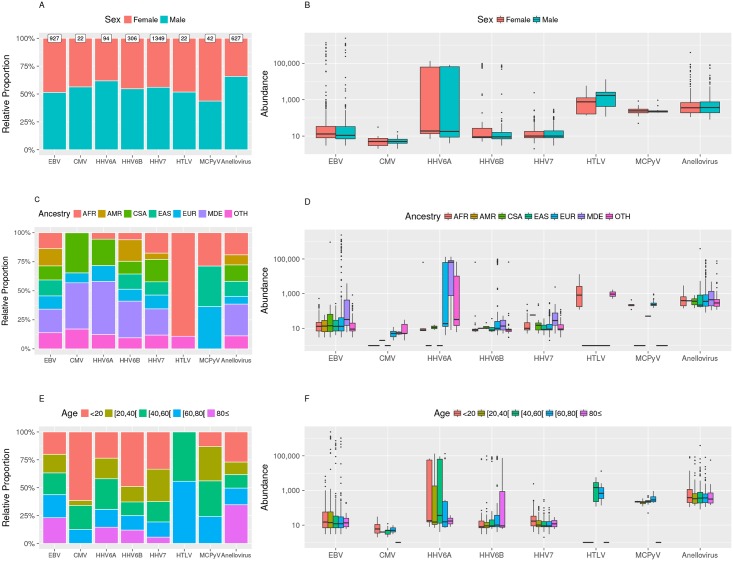
The characterization of the blood virome is important for the safety of blood-derived transfusion products, and for the identification of emerging pathogens. We explored non-human sequence data from whole-genome sequencing of blood from 8,240 individuals, none of whom were ascertained for any infectious disease. Viral sequences were extracted from the pool of sequence reads that did not map to the human reference genome. Analyses sifted through close to 1 Petabyte of sequence data and performed 0.5 trillion similarity searches. With a lower bound for identification of 2 viral genomes/100,000 cells, we mapped sequences to 94 different viruses, including sequences from 19 human DNA viruses, proviruses and RNA viruses (herpesviruses, anelloviruses, papillomaviruses, three polyomaviruses, adenovirus, HIV, HTLV, hepatitis B, hepatitis C, parvovirus B19, and influenza virus) in 42% of the study participants. Of possible relevance to transfusion medicine, we identified Merkel cell polyomavirus in 49 individuals, papillomavirus in blood of 13 individuals, parvovirus B19 in 6 individuals, and the presence of herpesvirus 8 in 3 individuals. The presence of DNA sequences from two RNA viruses was unexpected: Hepatitis C virus is revealing of an integration event, while the influenza virus sequence resulted from immunization with a DNA vaccine. Age, sex and ancestry contributed significantly to the prevalence of infection. The remaining 75 viruses mostly reflect extensive contamination of commercial reagents and from the environment. These technical problems represent a major challenge for the identification of novel human pathogens. Increasing availability of human whole-genome sequences will contribute substantial amounts of data on the composition of the normal and pathogenic human blood virome. Distinguishing contaminants from real human viruses is challenging.
Conflict of interest statement
Except ED, all authors are employees or own stock of Human Longevity Inc.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
