

cis-Regulatory Circuits Regulating NEK6 Kinase Overexpression in Transformed B Cells Are Super-Enhancer Independent
- PMID: 28329684
- PMCID: PMC5393904
- DOI: 10.1016/j.celrep.2017.02.067
cis-Regulatory Circuits Regulating NEK6 Kinase Overexpression in Transformed B Cells Are Super-Enhancer Independent
Abstract
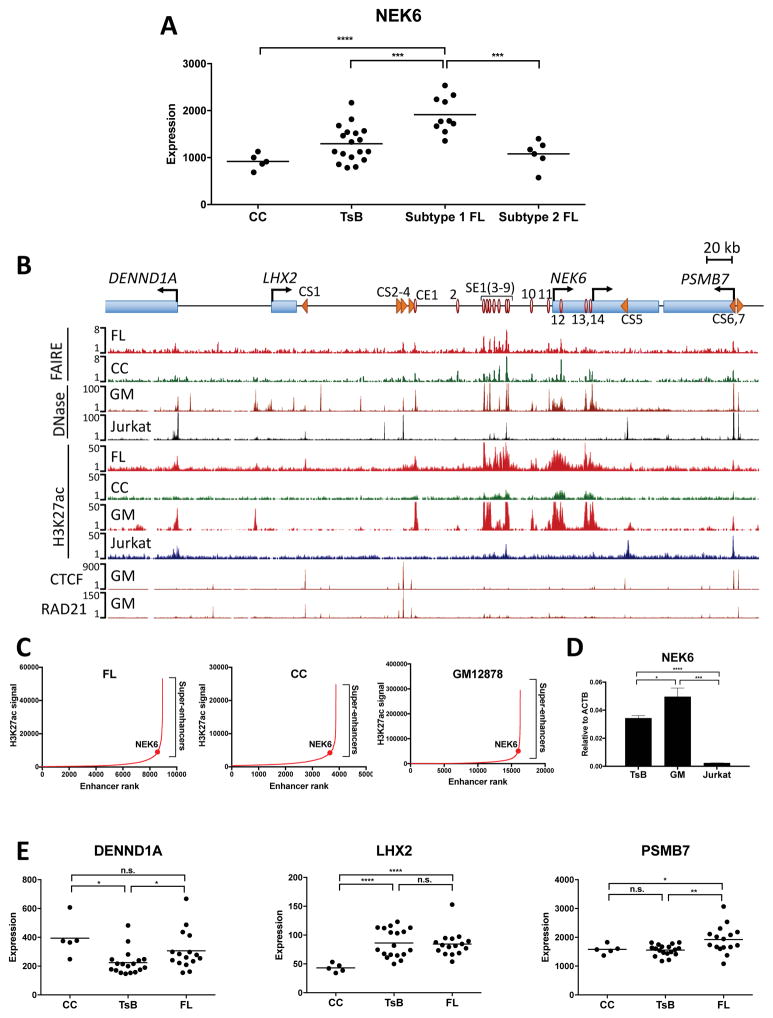
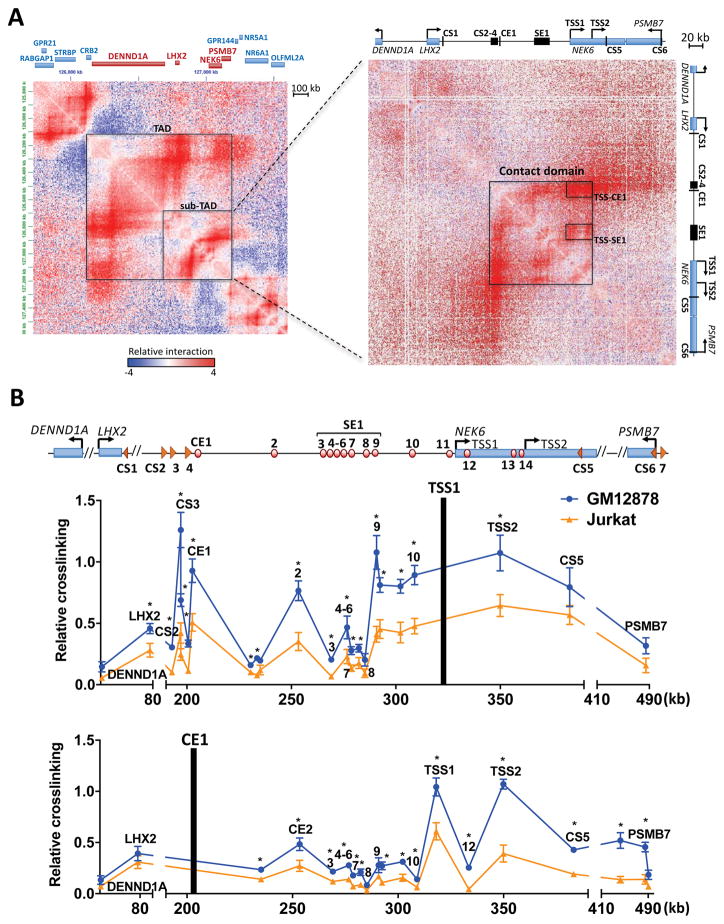
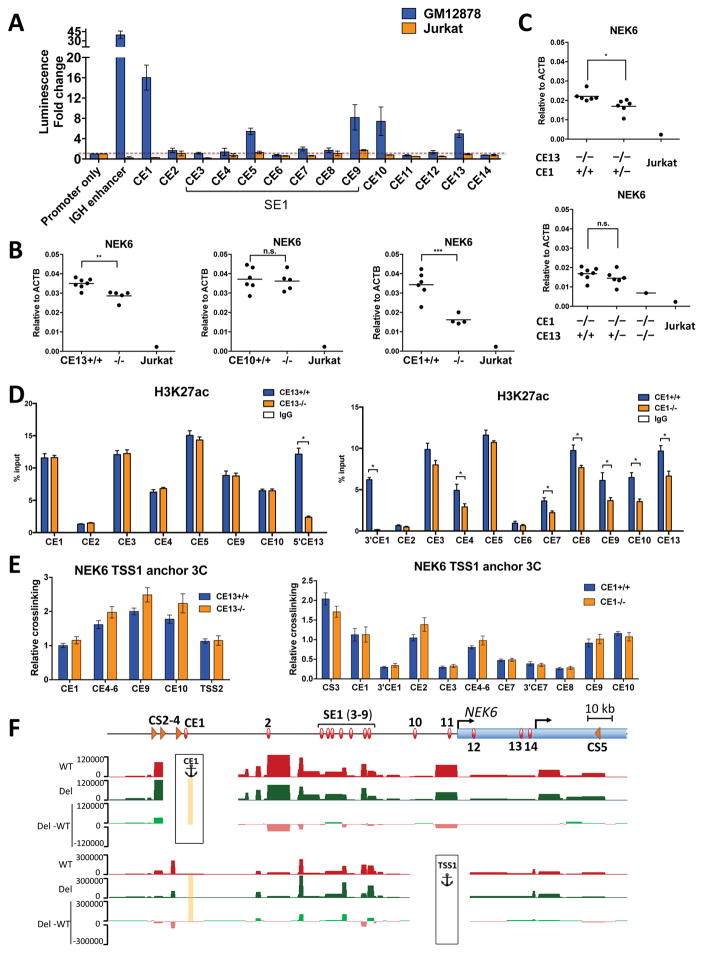
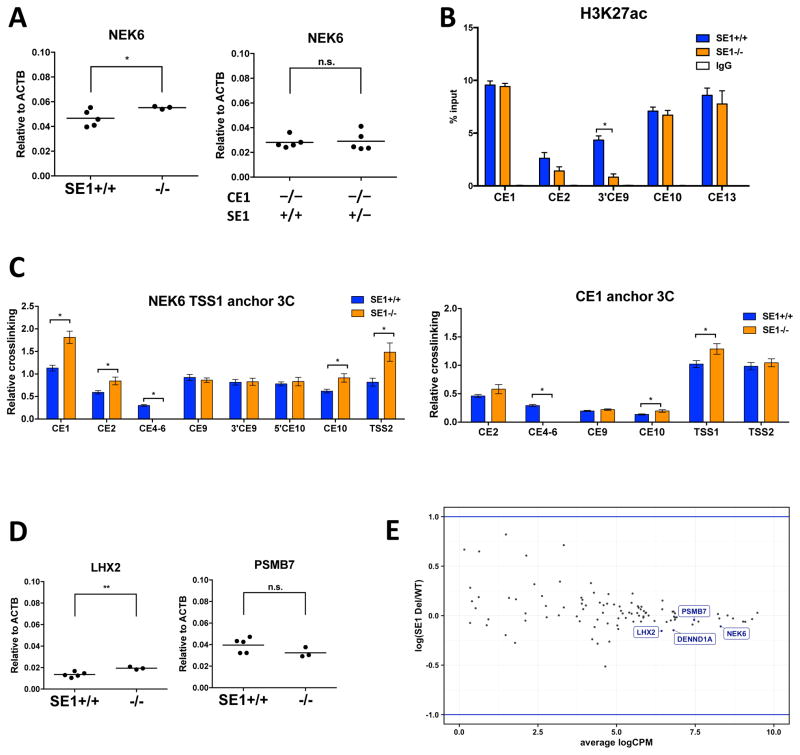
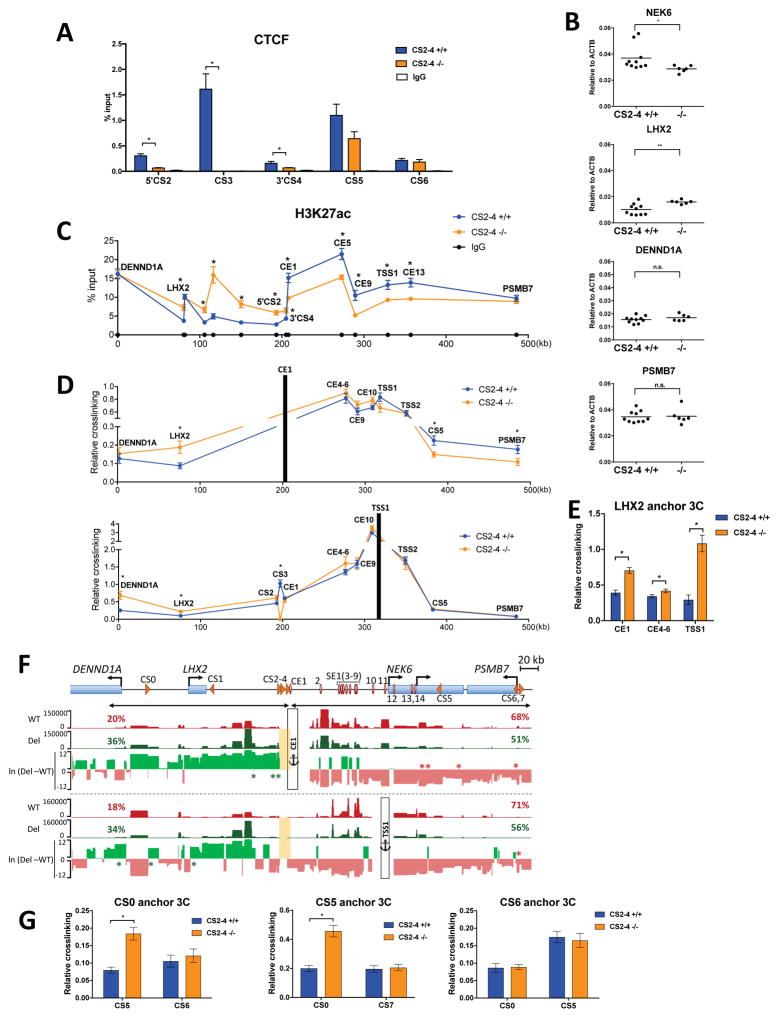
Alterations in distal regulatory elements that control gene expression underlie many diseases, including cancer. Epigenomic analyses of normal and diseased cells have produced correlative predictions for connections between dysregulated enhancers and target genes involved in pathogenesis. However, with few exceptions, these predicted cis-regulatory circuits remain untested. Here, we dissect cis-regulatory circuits that lead to overexpression of NEK6, a mitosis-associated kinase, in human B cell lymphoma. We find that only a minor subset of predicted enhancers is required for NEK6 expression. Indeed, an annotated super-enhancer is dispensable for NEK6 overexpression and for maintaining the architecture of a B cell-specific regulatory hub. A CTCF cluster serves as a chromatin and architectural boundary to block communication of the NEK6 regulatory hub with neighboring genes. Our findings emphasize that validation of predicted cis-regulatory circuits and super-enhancers is needed to prioritize transcriptional control elements as therapeutic targets.
Keywords: B cell lymphoma; NEK6; chromatin; chromosomal architecture; cis-regulatory circuits; gene regulation; super-enhancer.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
