

Functional materials discovery using energy-structure-function maps
- PMID: 28329756
- PMCID: PMC5458805
- DOI: 10.1038/nature21419
Functional materials discovery using energy-structure-function maps
Abstract
Molecular crystals cannot be designed in the same manner as macroscopic objects, because they do not assemble according to simple, intuitive rules. Their structures result from the balance of many weak interactions, rather than from the strong and predictable bonding patterns found in metal-organic frameworks and covalent organic frameworks. Hence, design strategies that assume a topology or other structural blueprint will often fail. Here we combine computational crystal structure prediction and property prediction to build energy-structure-function maps that describe the possible structures and properties that are available to a candidate molecule. Using these maps, we identify a highly porous solid, which has the lowest density reported for a molecular crystal so far. Both the structure of the crystal and its physical properties, such as methane storage capacity and guest-molecule selectivity, are predicted using the molecular structure as the only input. More generally, energy-structure-function maps could be used to guide the experimental discovery of materials with any target function that can be calculated from predicted crystal structures, such as electronic structure or mechanical properties.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
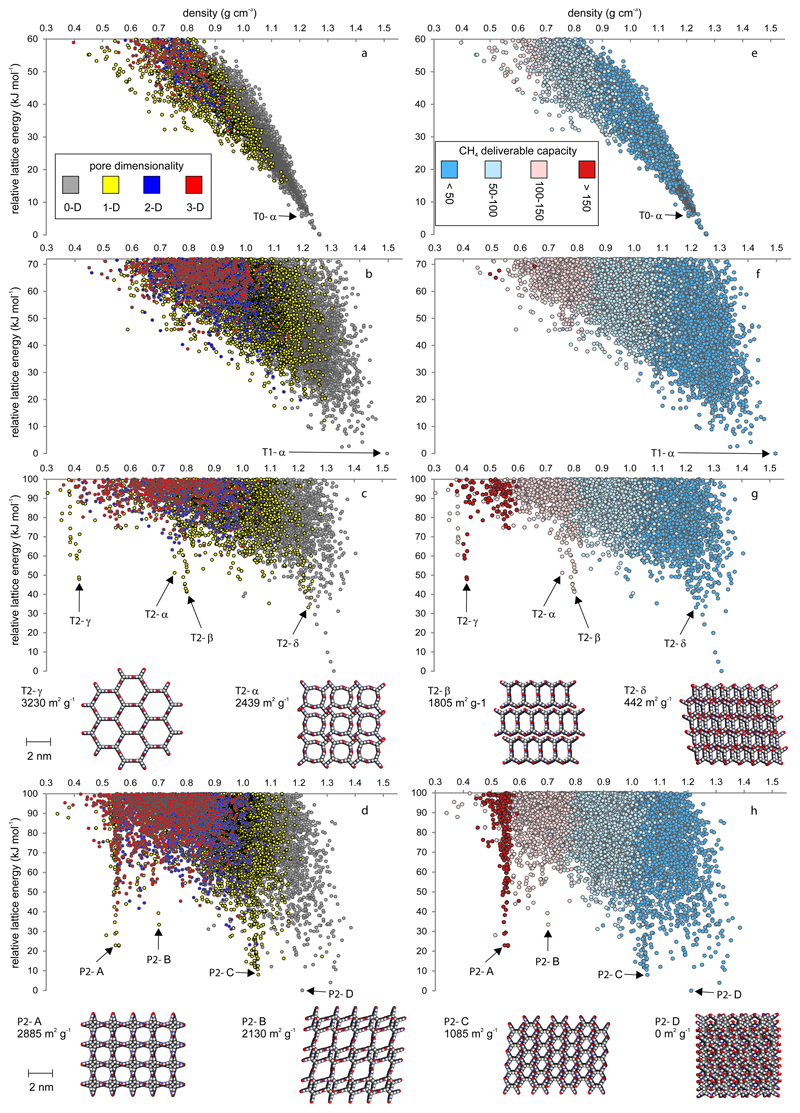
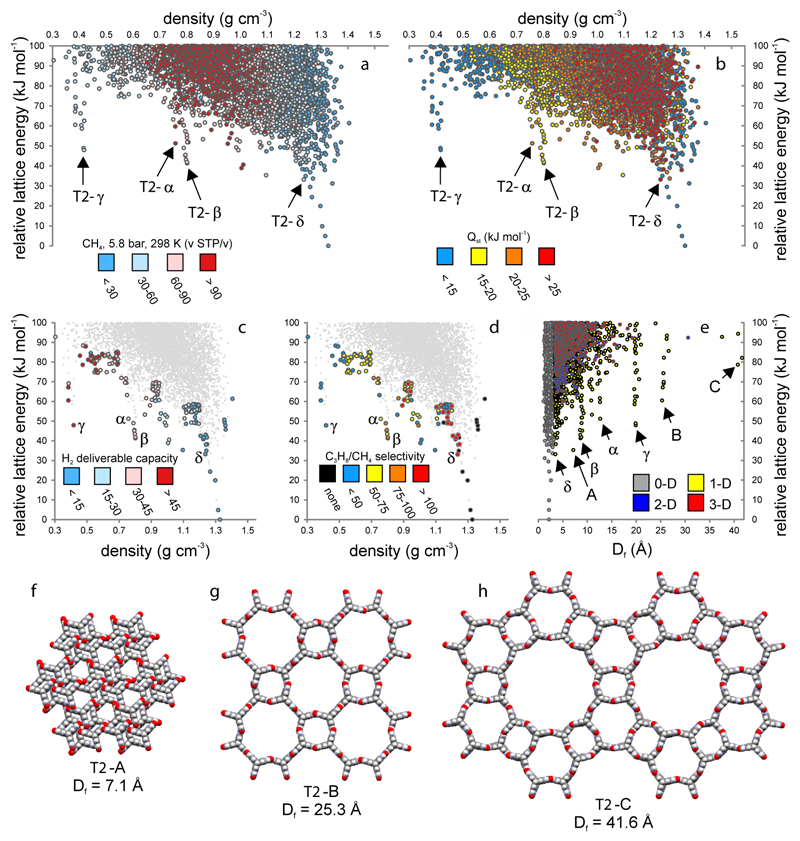
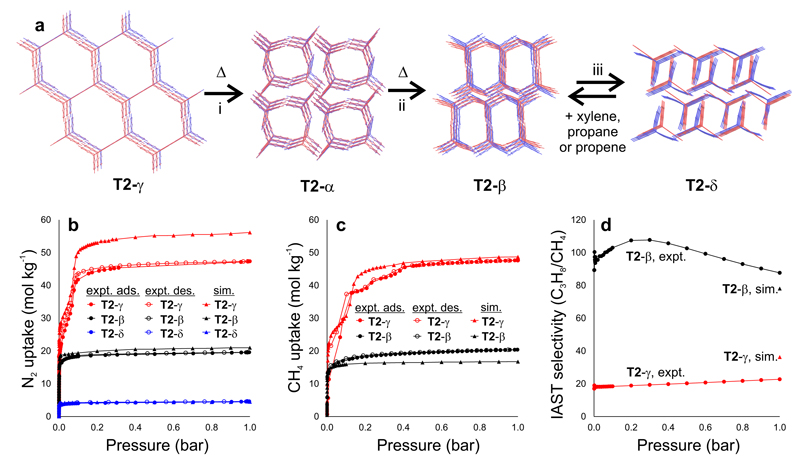
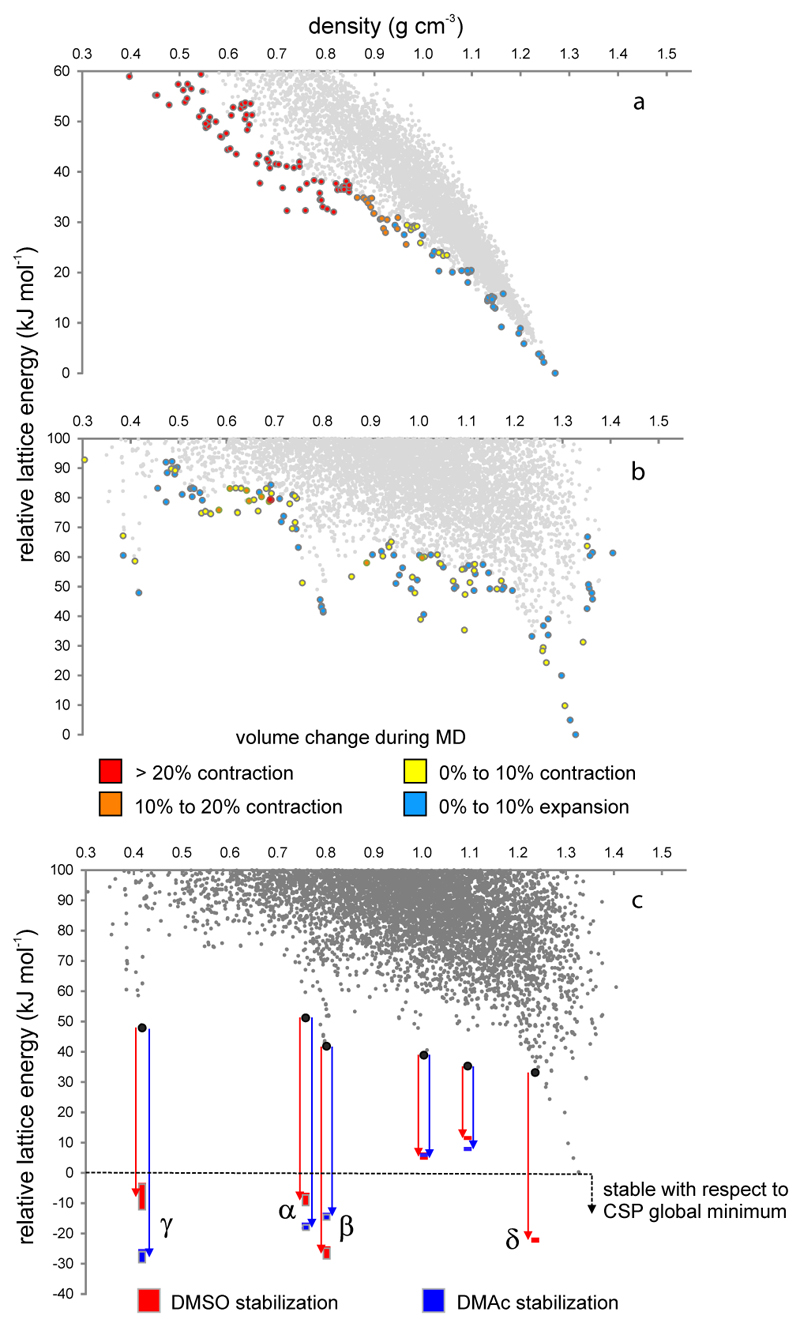
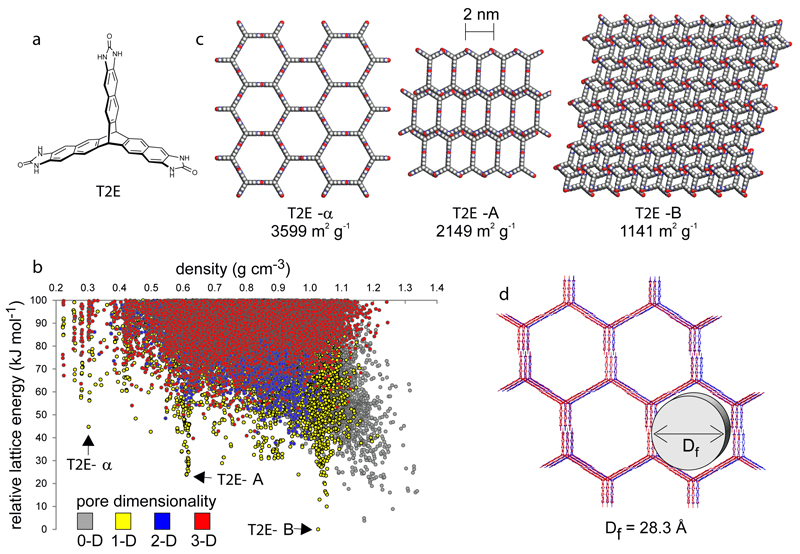
References
-
- Lewis DW, Willock DJ, Catlow CRA, Thomas JM, Hutchings GJ. De novo design of structure-directing agents for the synthesis of microporous solids. Nature. 1996;382:604–606.
-
- Oganov AR, et al. Ionic high-pressure form of elemental boron. Nature. 2009;457:863–867. - PubMed
-
- Ceder G, et al. Identification of cathode materials for lithium batteries guided by first-principles calculations. Nature. 1998;392:694–696.
-
- Marleny Rodriguez-Albelo L, et al. Zeolitic polyoxometalate-based metal–organic frameworks (Z-POMOFs): Computational evaluation of hypothetical polymorphs and the successful targeted synthesis of the redox-active Z-POMOF1. J Am Chem Soc. 2009;131:16078–16087. - PubMed
-
- Hachmann J, et al. The Harvard Clean Energy Project: Large-scale computational screening and design of organic photovoltaics on the World Community Grid. J Phys Chem Lett. 2011;2:2241–2251.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
