Somatic mutations reveal asymmetric cellular dynamics in the early human embryo
- PMID: 28329761
- PMCID: PMC6169740
- DOI: 10.1038/nature21703
Somatic mutations reveal asymmetric cellular dynamics in the early human embryo
Abstract
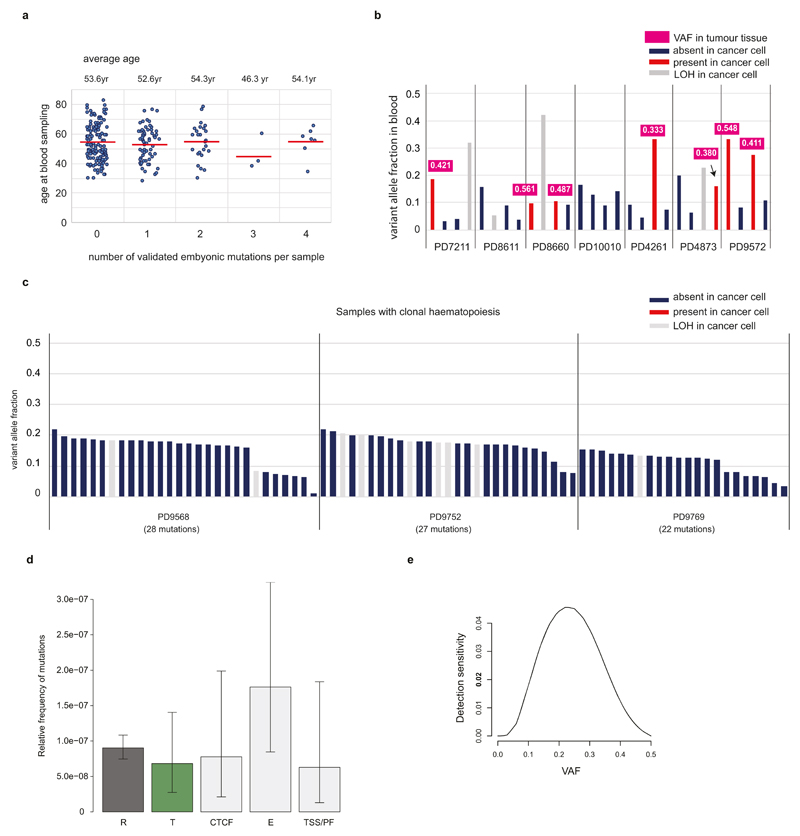
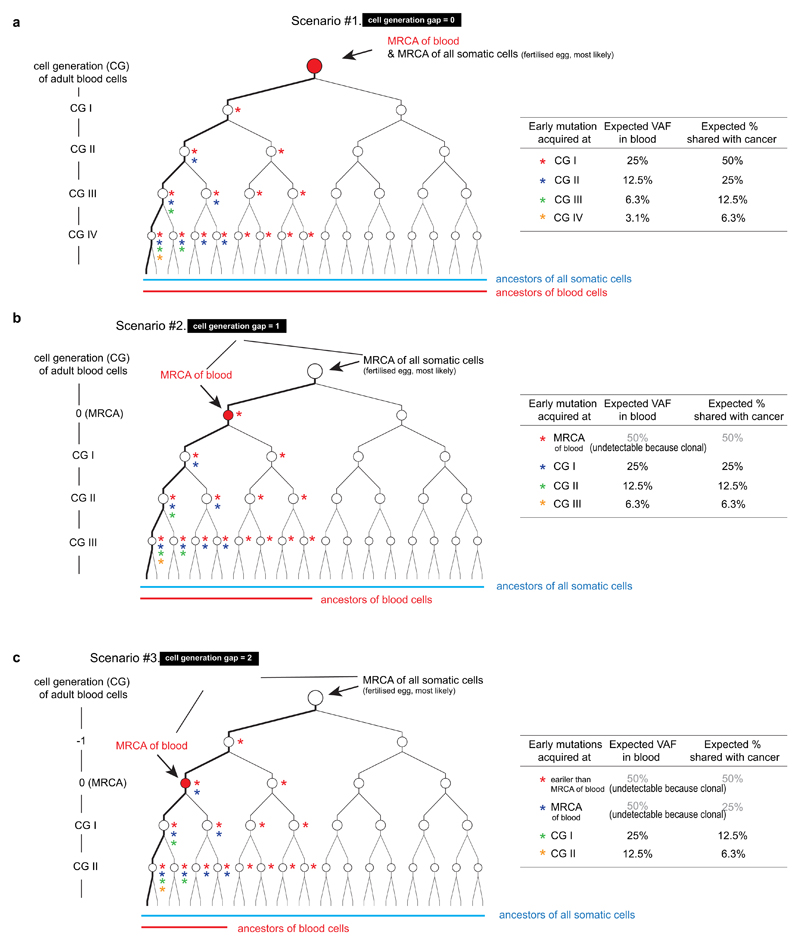
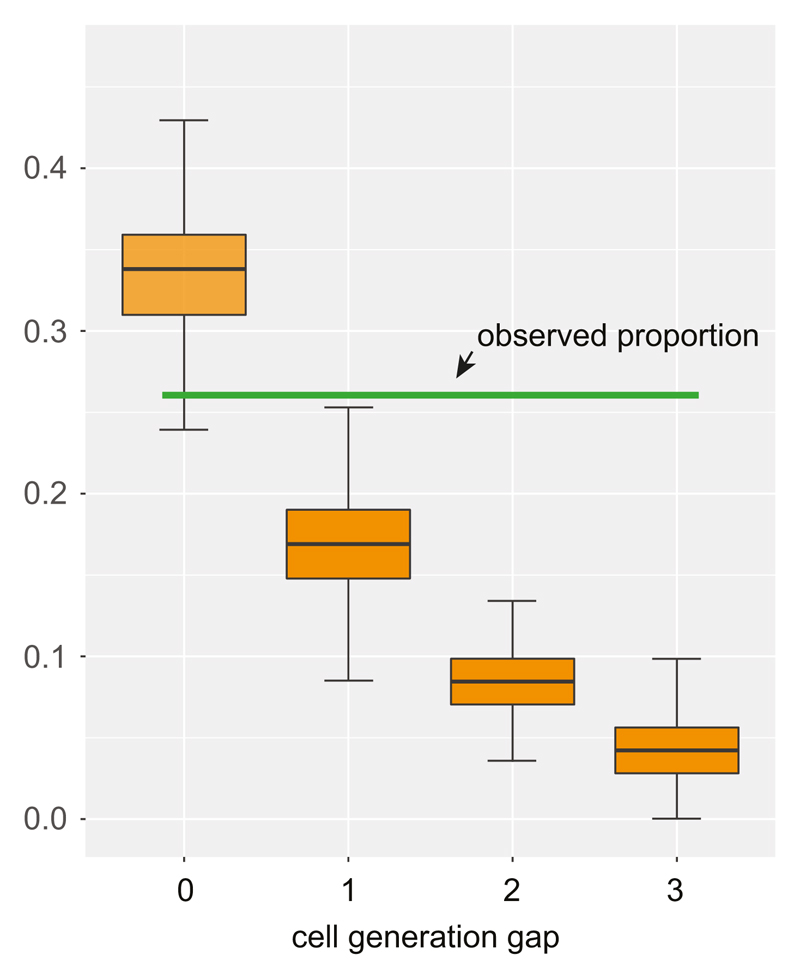
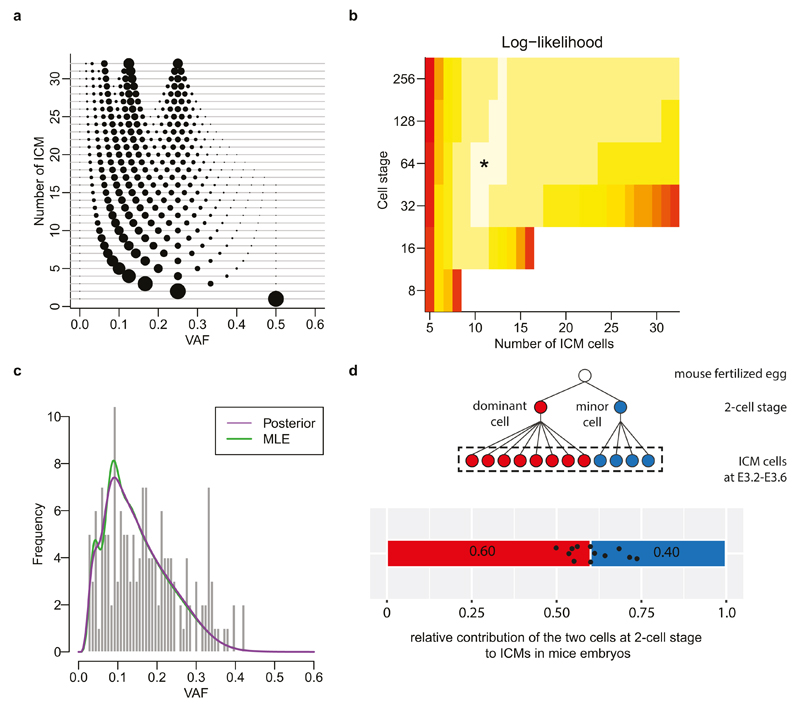
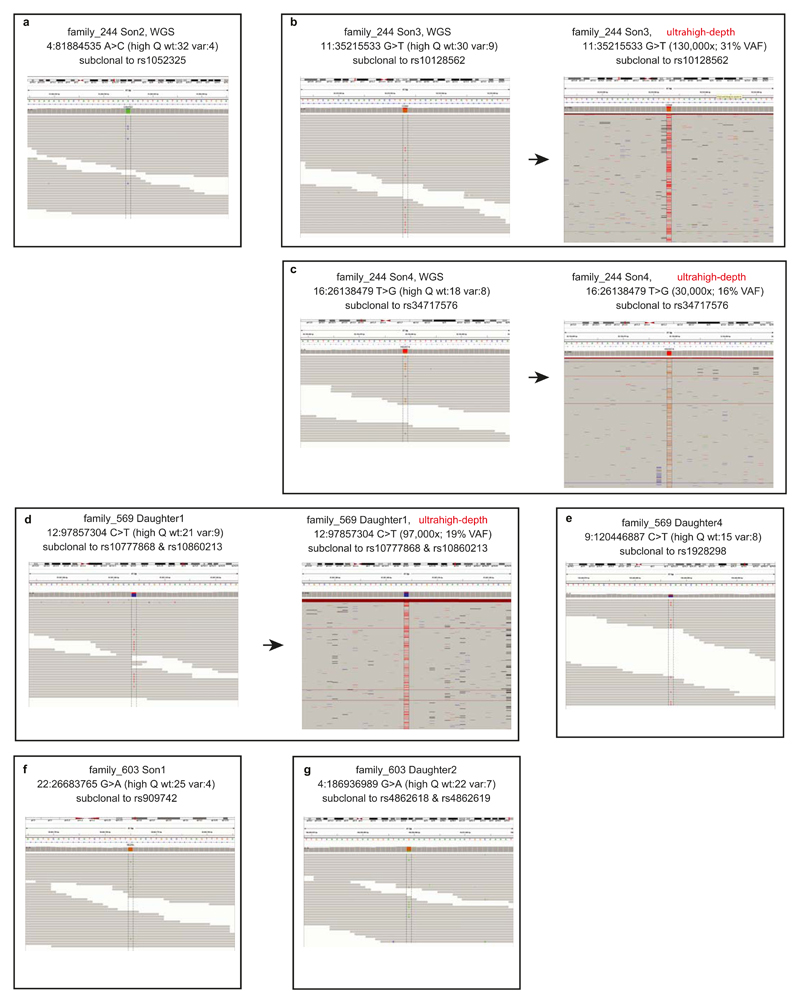
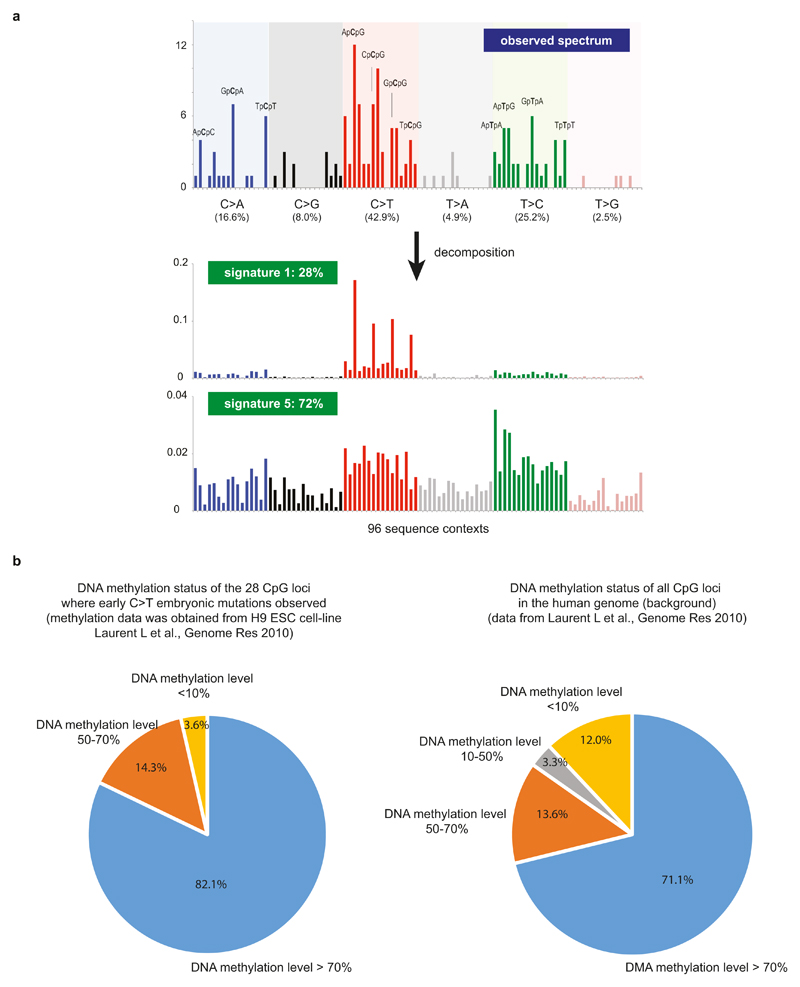
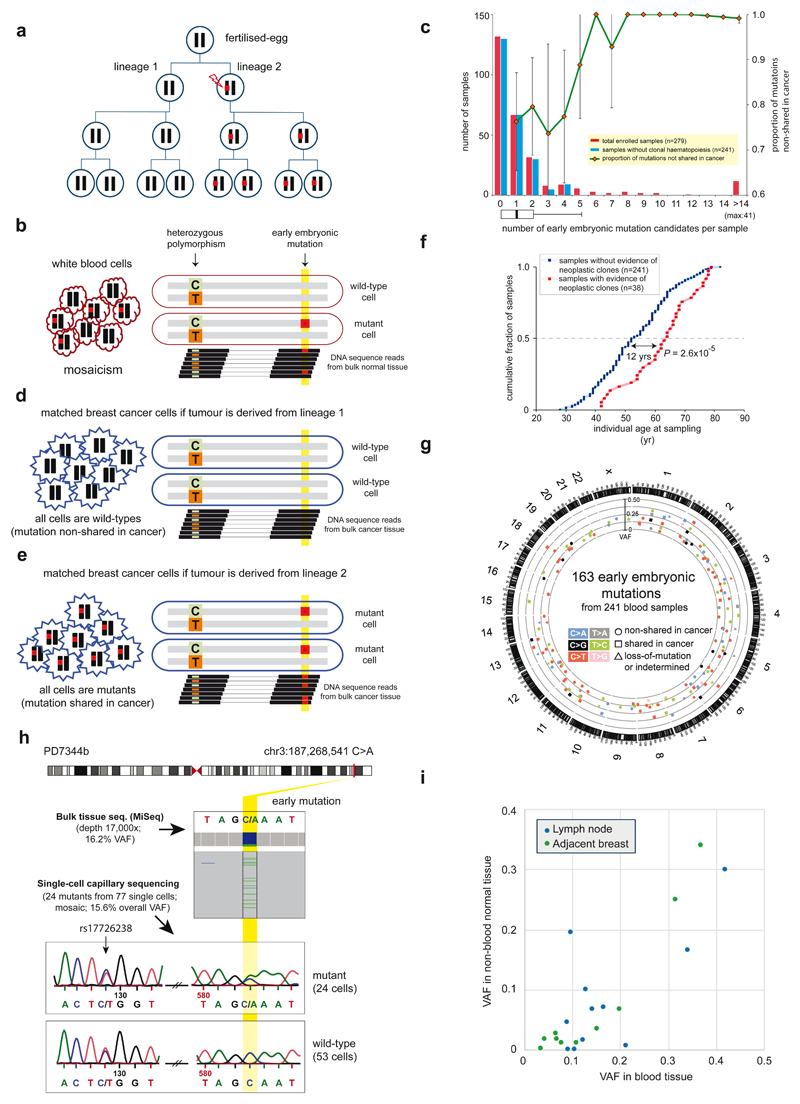
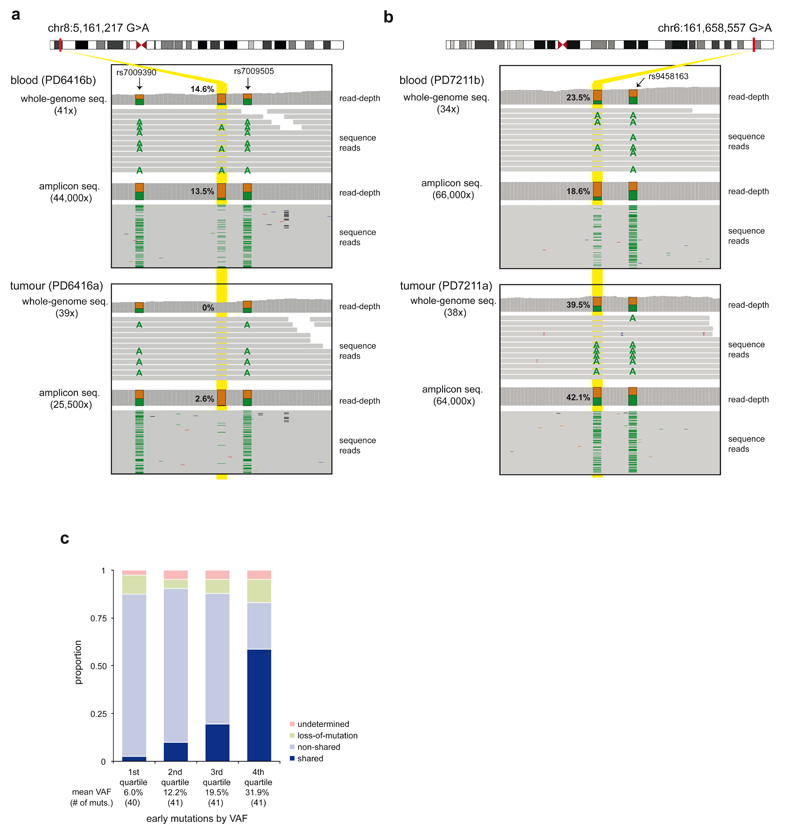
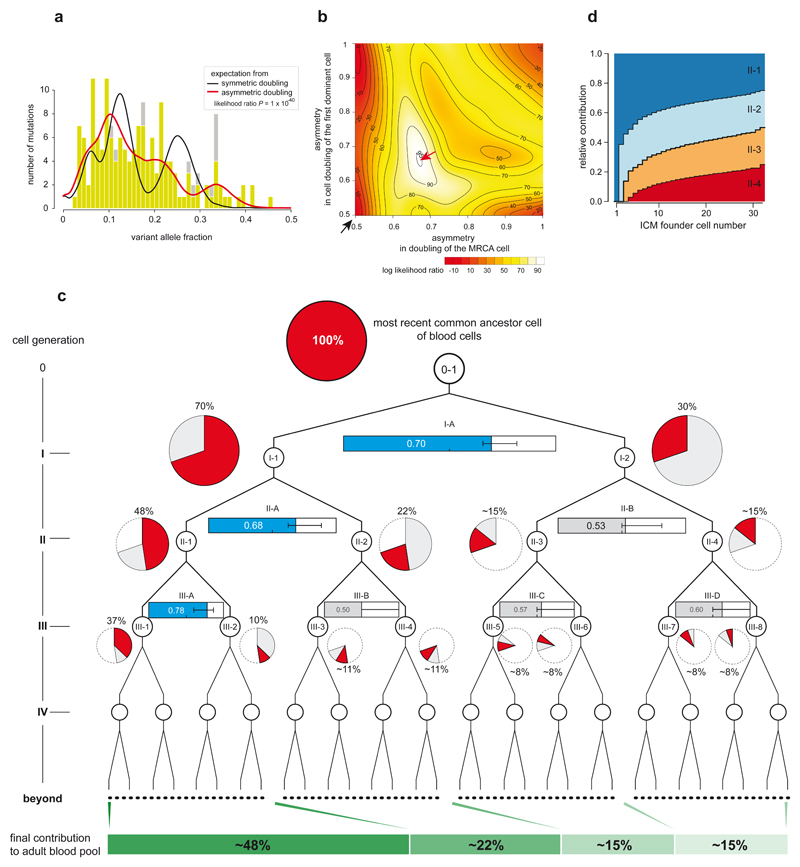
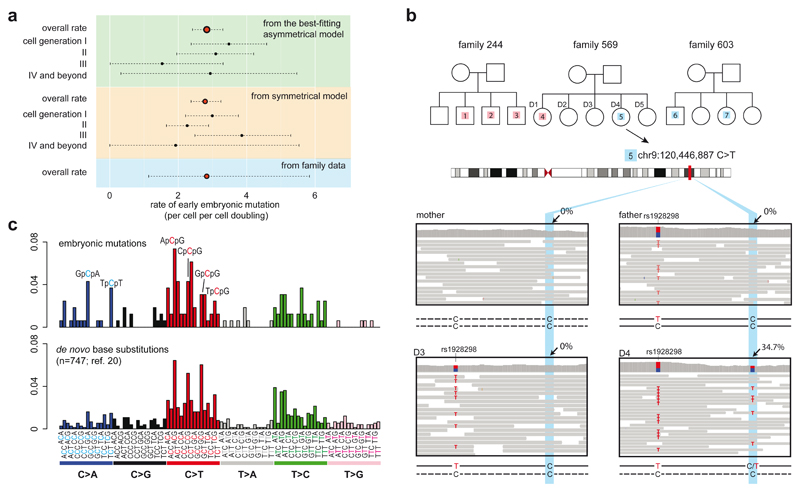
Somatic cells acquire mutations throughout the course of an individual's life. Mutations occurring early in embryogenesis are often present in a substantial proportion of, but not all, cells in postnatal humans and thus have particular characteristics and effects. Depending on their location in the genome and the proportion of cells they are present in, these mosaic mutations can cause a wide range of genetic disease syndromes and predispose carriers to cancer. They have a high chance of being transmitted to offspring as de novo germline mutations and, in principle, can provide insights into early human embryonic cell lineages and their contributions to adult tissues. Although it is known that gross chromosomal abnormalities are remarkably common in early human embryos, our understanding of early embryonic somatic mutations is very limited. Here we use whole-genome sequences of normal blood from 241 adults to identify 163 early embryonic mutations. We estimate that approximately three base substitution mutations occur per cell per cell-doubling event in early human embryogenesis and these are mainly attributable to two known mutational signatures. We used the mutations to reconstruct developmental lineages of adult cells and demonstrate that the two daughter cells of many early embryonic cell-doubling events contribute asymmetrically to adult blood at an approximately 2:1 ratio. This study therefore provides insights into the mutation rates, mutational processes and developmental outcomes of cell dynamics that operate during early human embryogenesis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures