

A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis
- PMID: 28330499
- PMCID: PMC5363043
- DOI: 10.1186/s13073-017-0417-1
A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis
Abstract
Background: Systemic sclerosis (SSc) is a multi-organ autoimmune disease characterized by skin fibrosis. Internal organ involvement is heterogeneous. It is unknown whether disease mechanisms are common across all involved affected tissues or if each manifestation has a distinct underlying pathology.
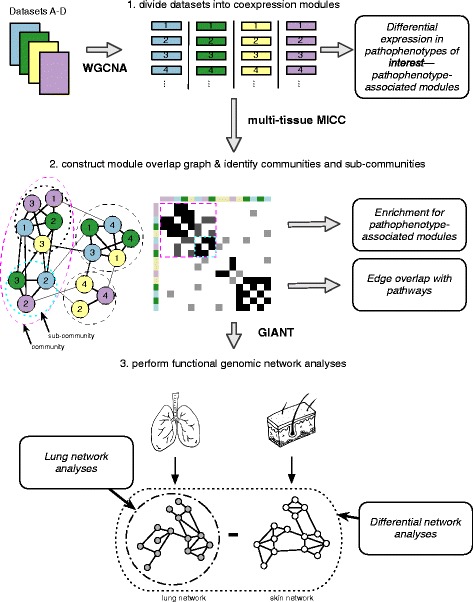
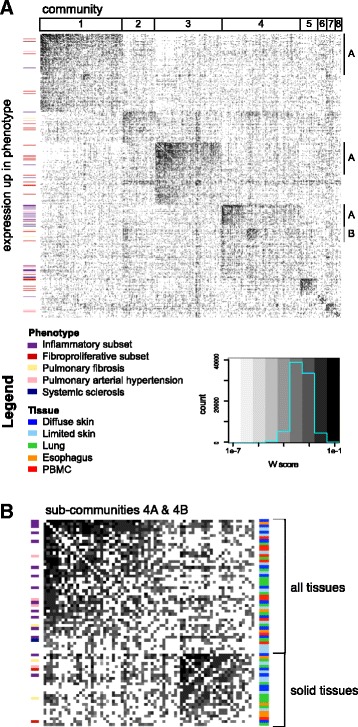
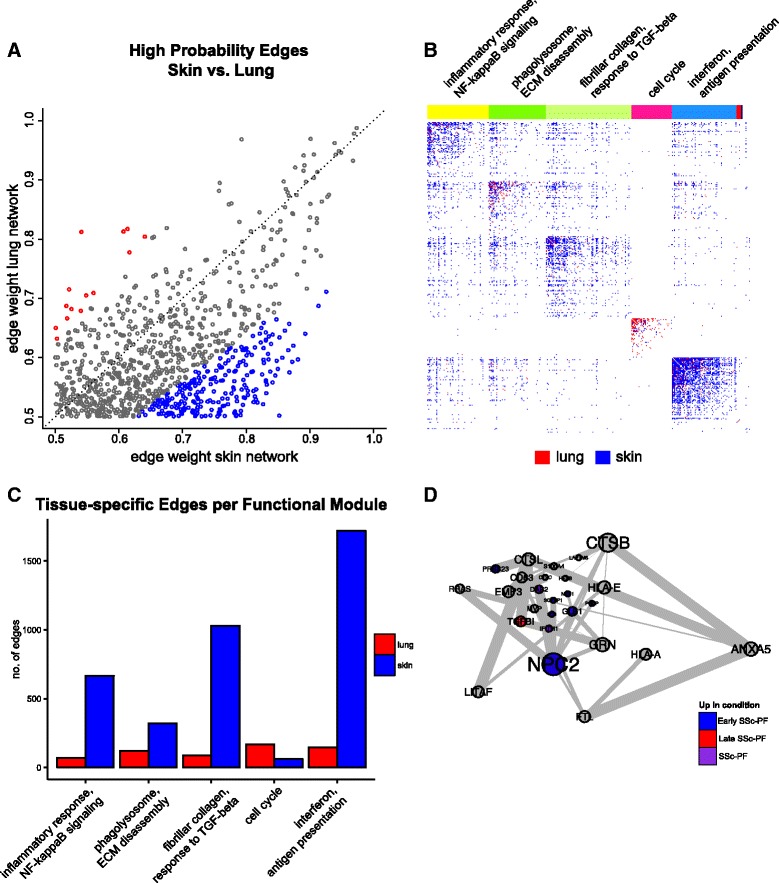
Methods: We used consensus clustering to compare gene expression profiles of biopsies from four SSc-affected tissues (skin, lung, esophagus, and peripheral blood) from patients with SSc, and the related conditions pulmonary fibrosis (PF) and pulmonary arterial hypertension, and derived a consensus disease-associate signature across all tissues. We used this signature to query tissue-specific functional genomic networks. We performed novel network analyses to contrast the skin and lung microenvironments and to assess the functional role of the inflammatory and fibrotic genes in each organ. Lastly, we tested the expression of macrophage activation state-associated gene sets for enrichment in skin and lung using a Wilcoxon rank sum test.
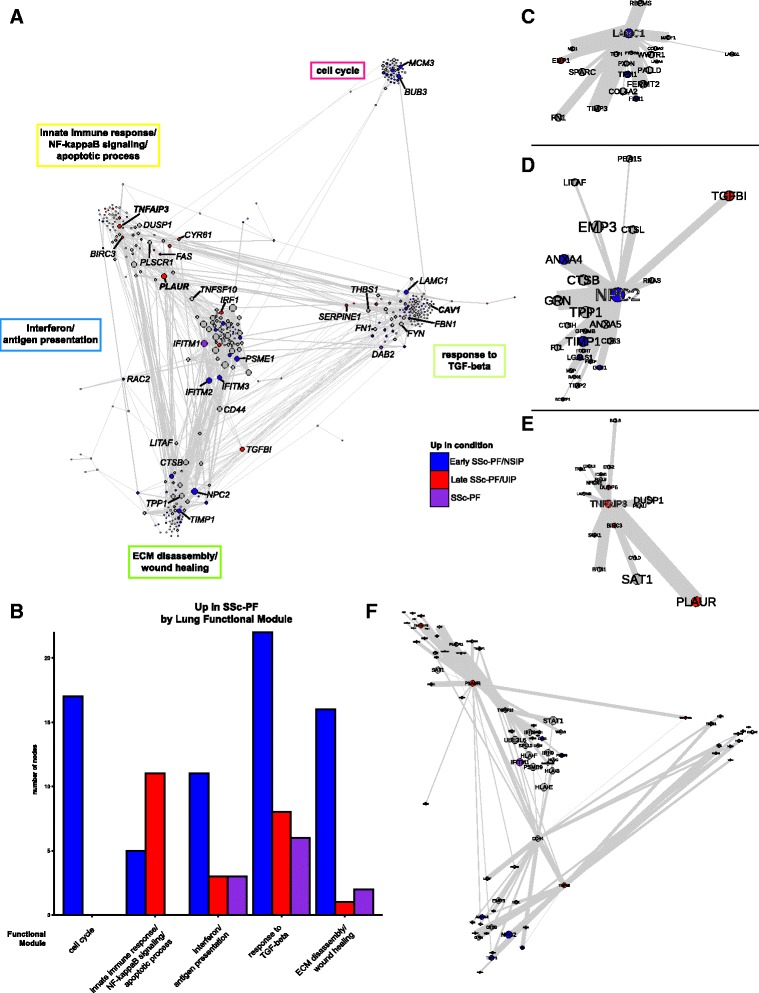
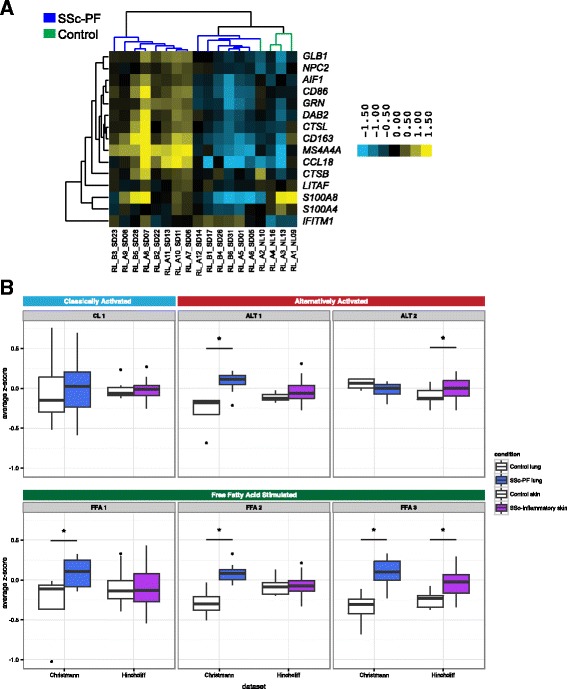
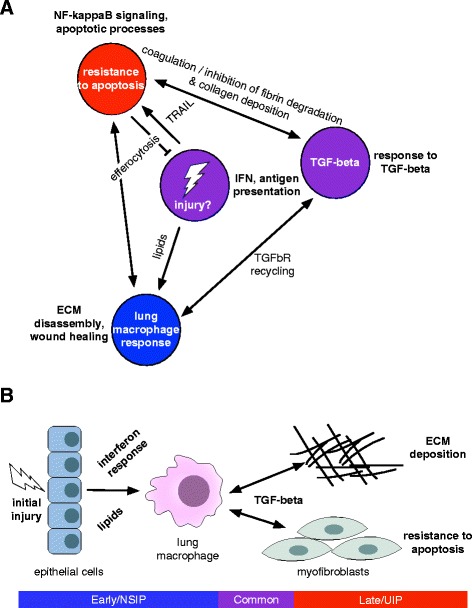
Results: We identified a common pathogenic gene expression signature-an immune-fibrotic axis-indicative of pro-fibrotic macrophages (MØs) in multiple tissues (skin, lung, esophagus, and peripheral blood mononuclear cells) affected by SSc. While the co-expression of these genes is common to all tissues, the functional consequences of this upregulation differ by organ. We used this disease-associated signature to query tissue-specific functional genomic networks to identify common and tissue-specific pathologies of SSc and related conditions. In contrast to skin, in the lung-specific functional network we identify a distinct lung-resident MØ signature associated with lipid stimulation and alternative activation. In keeping with our network results, we find distinct MØ alternative activation transcriptional programs in SSc-associated PF lung and in the skin of patients with an "inflammatory" SSc gene expression signature.
Conclusions: Our results suggest that the innate immune system is central to SSc disease processes but that subtle distinctions exist between tissues. Our approach provides a framework for examining molecular signatures of disease in fibrosis and autoimmune diseases and for leveraging publicly available data to understand common and tissue-specific disease processes in complex human diseases.
Keywords: Functional genomics; Lung disease; Macrophage; Scleroderma; Systemic sclerosis.
Figures
References
-
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, Chasman DI, FitzGerald GA, Dolinski K, Grosser T, and Troyanskaya OG. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47:569–576. - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
