

Quantitative Tyrosine Phosphoproteomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor-treated Lung Adenocarcinoma Cells Reveals Potential Novel Biomarkers of Therapeutic Response
- PMID: 28331001
- PMCID: PMC5417828
- DOI: 10.1074/mcp.M117.067439
Quantitative Tyrosine Phosphoproteomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor-treated Lung Adenocarcinoma Cells Reveals Potential Novel Biomarkers of Therapeutic Response
Abstract
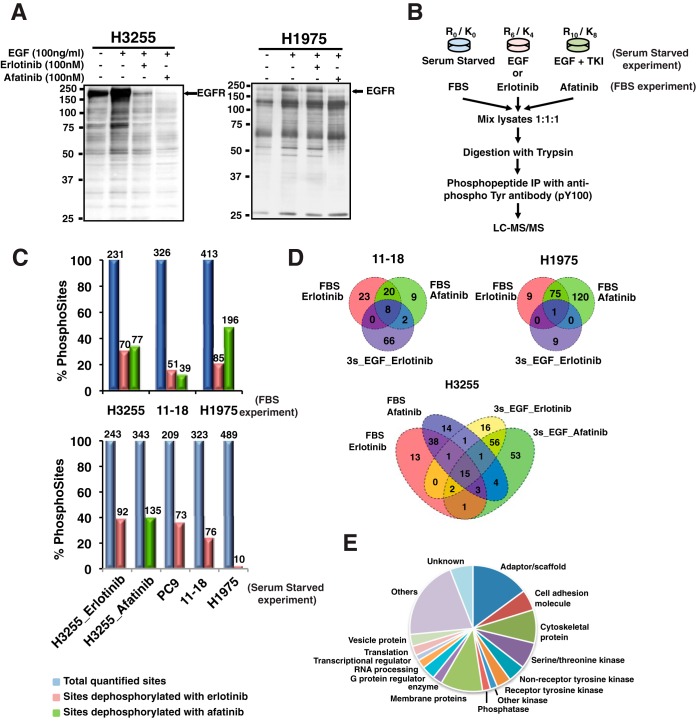
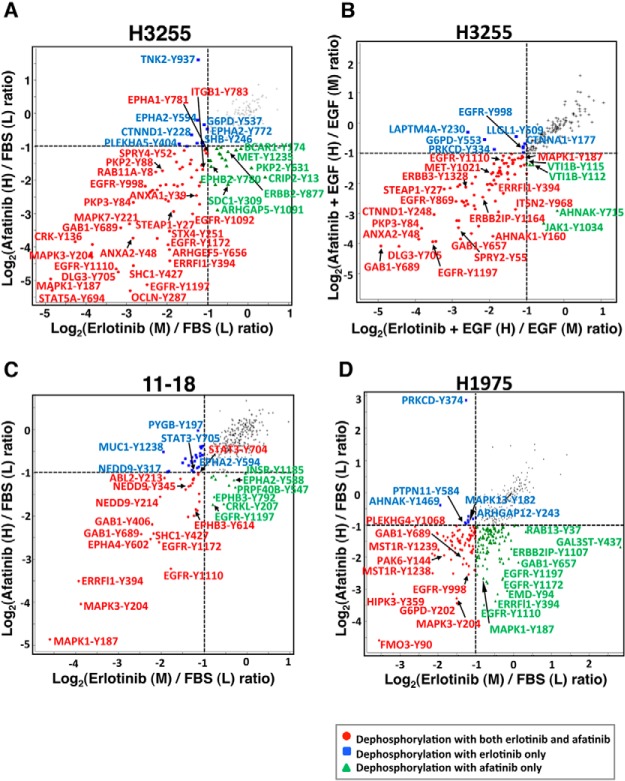
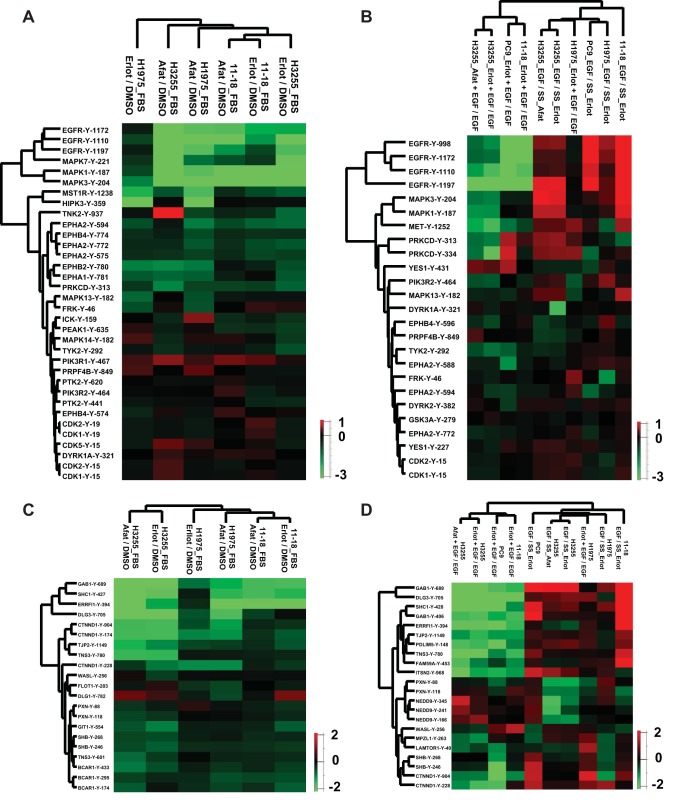
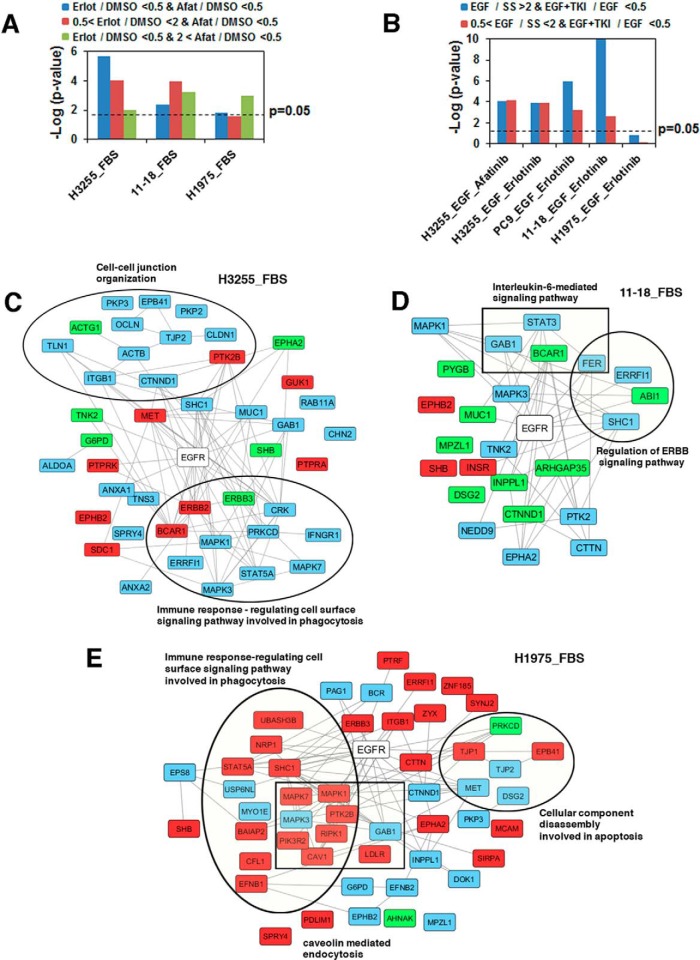
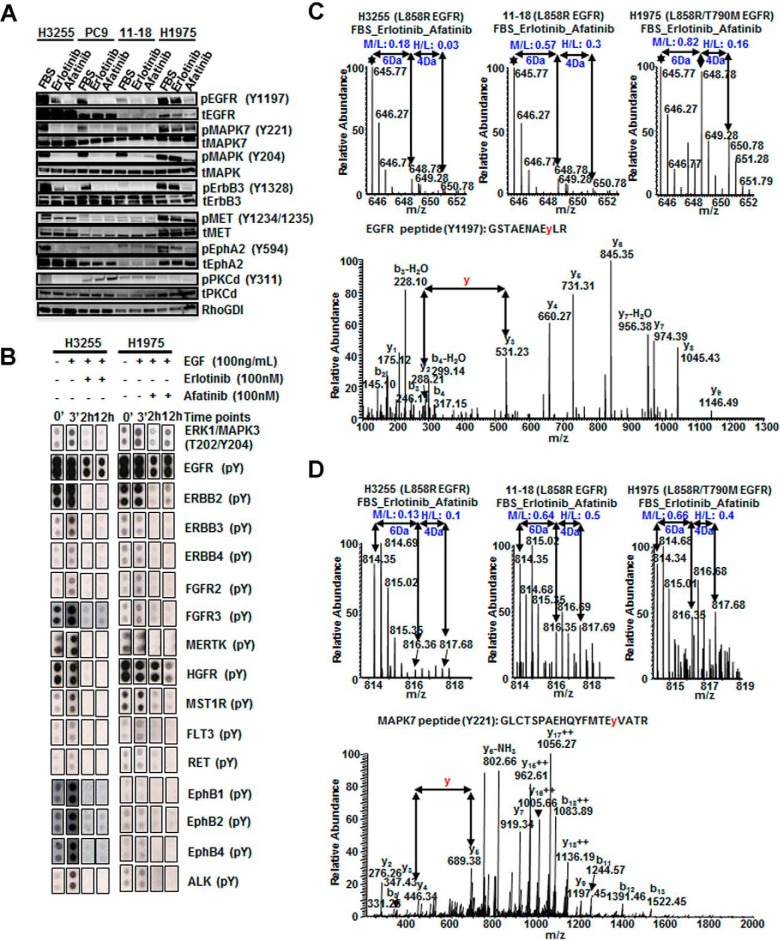
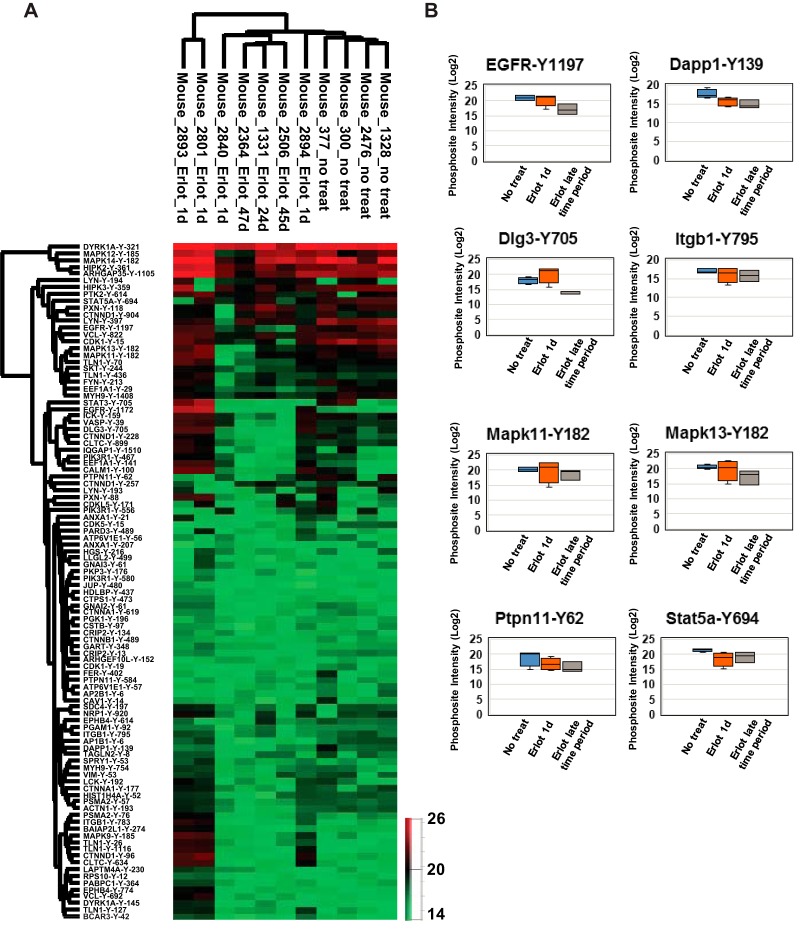
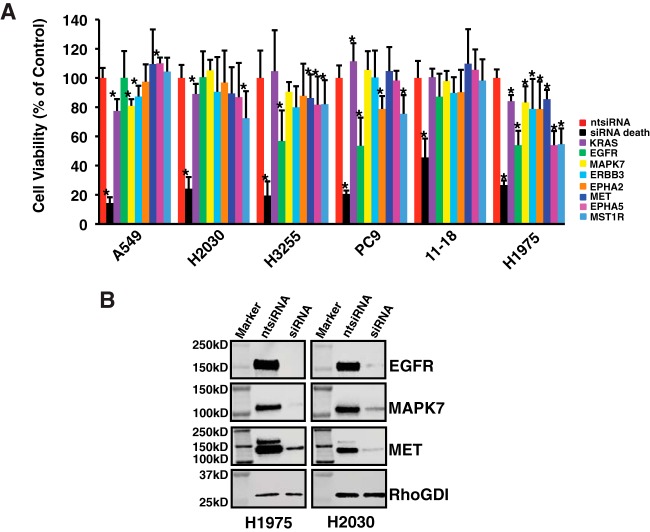
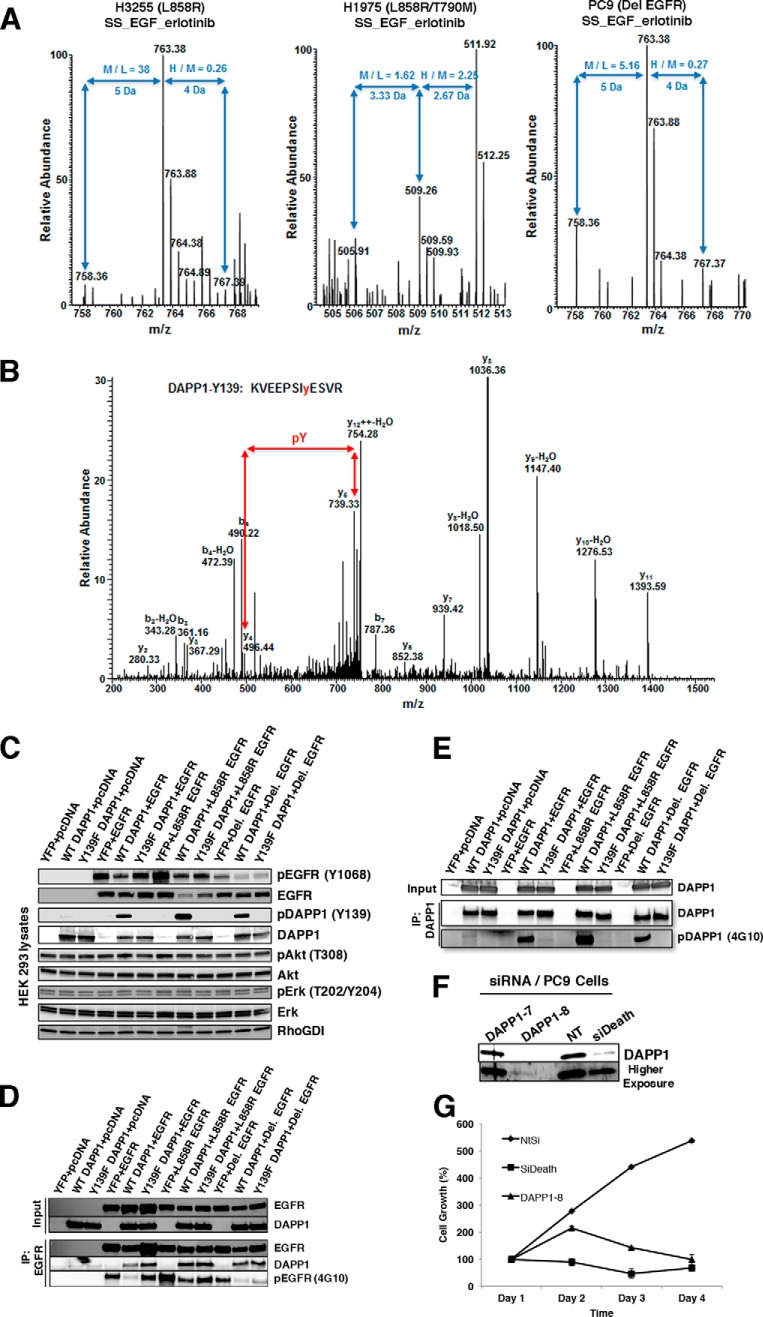
Mutations in the Epidermal growth factor receptor (EGFR) kinase domain, such as the L858R missense mutation and deletions spanning the conserved sequence 747LREA750, are sensitive to tyrosine kinase inhibitors (TKIs). The gatekeeper site residue mutation, T790M accounts for around 60% of acquired resistance to EGFR TKIs. The first generation EGFR TKIs, erlotinib and gefitinib, and the second generation inhibitor, afatinib are FDA approved for initial treatment of EGFR mutated lung adenocarcinoma. The predominant biomarker of EGFR TKI responsiveness is the presence of EGFR TKI-sensitizing mutations. However, 30-40% of patients with EGFR mutations exhibit primary resistance to these TKIs, underscoring the unmet need of identifying additional biomarkers of treatment response. Here, we sought to characterize the dynamics of tyrosine phosphorylation upon EGFR TKI treatment of mutant EGFR-driven human lung adenocarcinoma cell lines with varying sensitivity to EGFR TKIs, erlotinib and afatinib. We employed stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative mass spectrometry to identify and quantify tyrosine phosphorylated peptides. The proportion of tyrosine phosphorylated sites that had reduced phosphorylation upon erlotinib or afatinib treatment correlated with the degree of TKI-sensitivity. Afatinib, an irreversible EGFR TKI, more effectively inhibited tyrosine phosphorylation of a majority of the substrates. The phosphosites with phosphorylation SILAC ratios that correlated with the TKI-sensitivity of the cell lines include sites on kinases, such as EGFR-Y1197 and MAPK7-Y221, and adaptor proteins, such as SHC1-Y349/350, ERRFI1-Y394, GAB1-Y689, STAT5A-Y694, DLG3-Y705, and DAPP1-Y139, suggesting these are potential biomarkers of TKI sensitivity. DAPP1, is a novel target of mutant EGFR signaling and Y-139 is the major site of DAPP1 tyrosine phosphorylation. We also uncovered several off-target effects of these TKIs, such as MST1R-Y1238/Y1239 and MET-Y1252/1253. This study provides unique insight into the TKI-mediated modulation of mutant EGFR signaling, which can be applied to the development of biomarkers of EGFR TKI response.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
We have declared no conflicts of interest
Figures
References
-
- Siegel R., Naishadham D., and Jemal A. (2013) Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 - PubMed
-
- Dowell J., Minna J. D., and Kirkpatrick P. (2005) Erlotinib hydrochloride. Nat. Rev. Drug Discov. 4, 13–14 - PubMed
-
- Melnikova I., and Golden J. (2004) Targeting protein kinases. Nat. Rev. Drug Discov. 3, 993–994 - PubMed
-
- Sharma S. P. (2007) New predictors of survival for early-stage NSCLC. Lancet Oncol. 8, 288 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
