

Multiple Sources of Genetic Diversity of Influenza A Viruses during the Hajj
- PMID: 28331081
- PMCID: PMC5432881
- DOI: 10.1128/JVI.00096-17
Multiple Sources of Genetic Diversity of Influenza A Viruses during the Hajj
Abstract
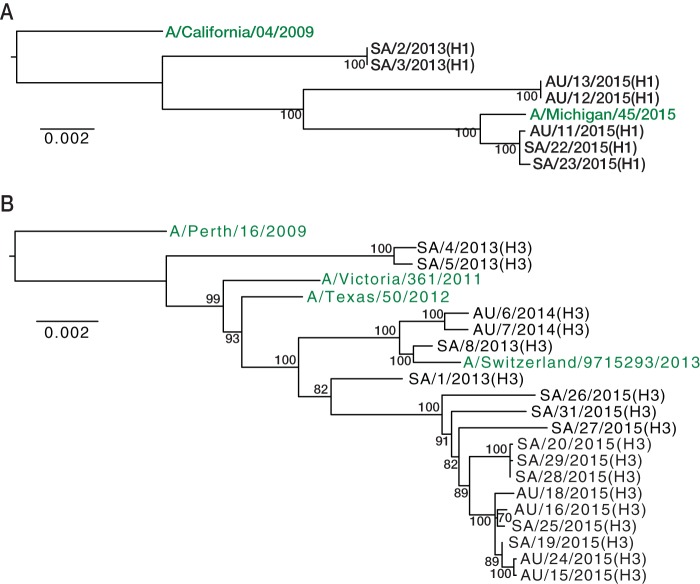
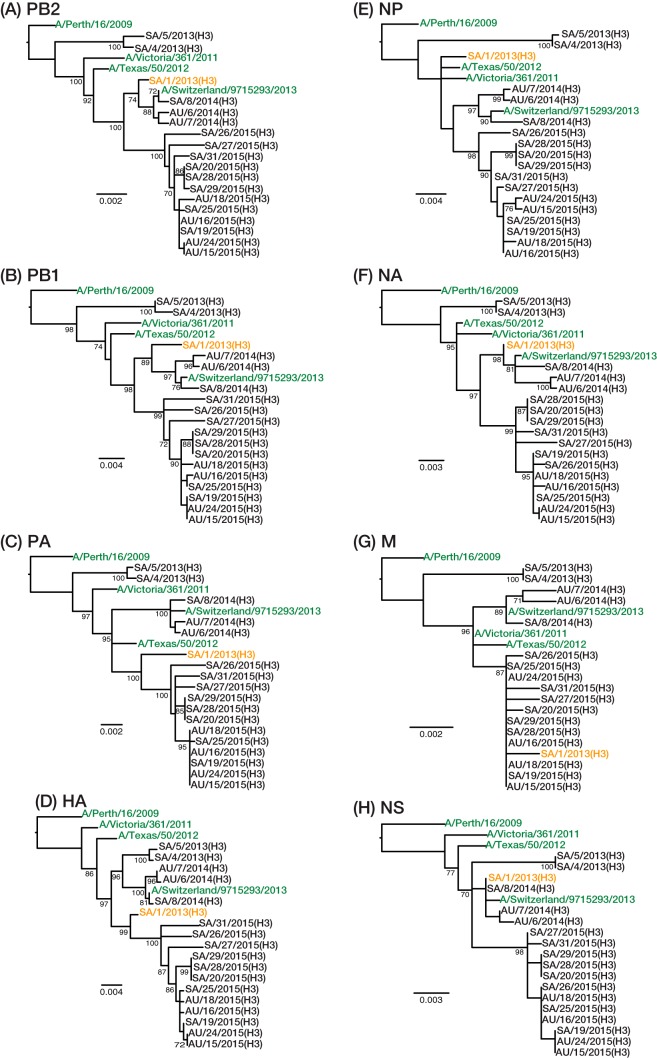
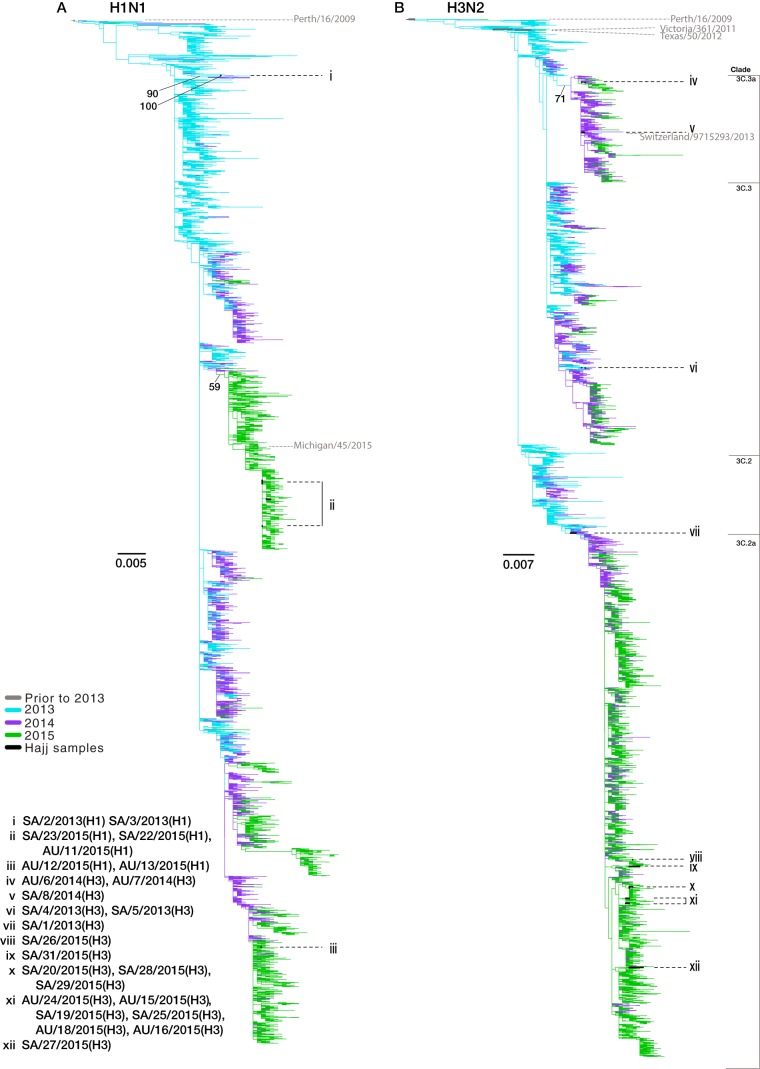
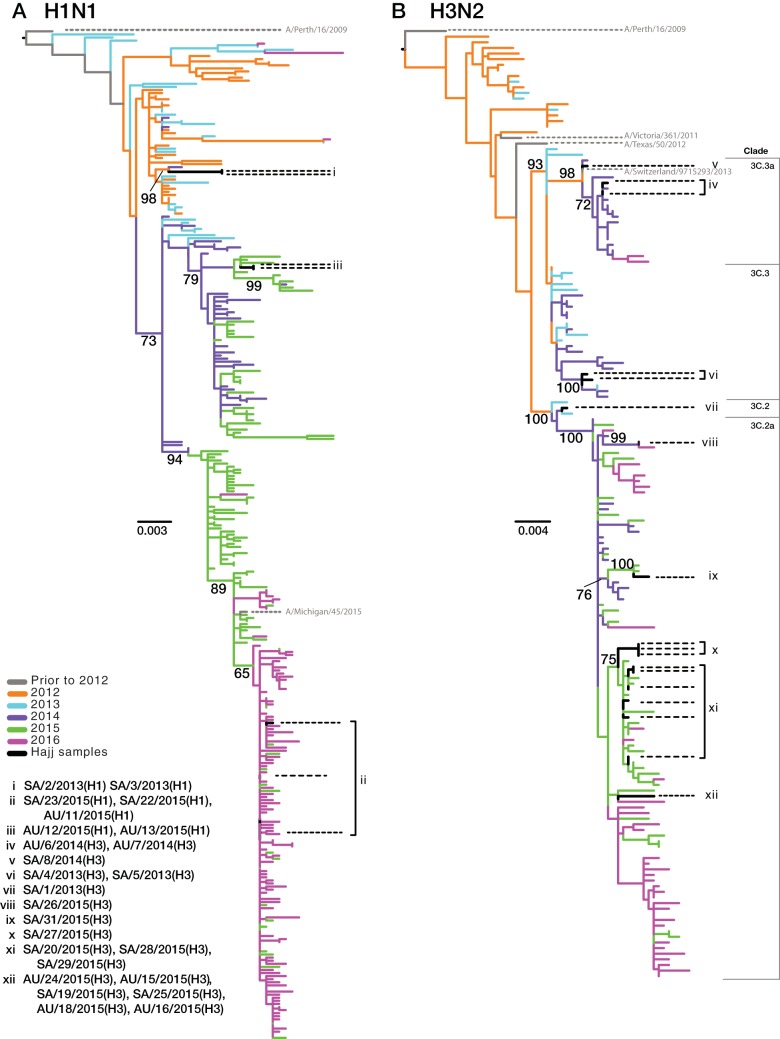
Outbreaks of respiratory virus infection at mass gatherings pose significant health risks to attendees, host communities, and ultimately the global population if they help facilitate viral emergence. However, little is known about the genetic diversity, evolution, and patterns of viral transmission during mass gatherings, particularly how much diversity is generated by in situ transmission compared to that imported from other locations. Here, we describe the genome-scale evolution of influenza A viruses sampled from the Hajj pilgrimages at Makkah during 2013 to 2015. Phylogenetic analysis revealed that the diversity of influenza viruses at the Hajj pilgrimages was shaped by multiple introduction events, comprising multiple cocirculating lineages in each year, including those that have circulated in the Middle East and those whose origins likely lie on different continents. At the scale of individual hosts, the majority of minor variants resulted from de novo mutation, with only limited evidence of minor variant transmission or minor variants circulating at subconsensus level despite the likely identification of multiple transmission clusters. Together, these data highlight the complexity of influenza virus infection at the Hajj pilgrimages, reflecting a mix of global genetic diversity drawn from multiple sources combined with local transmission, and reemphasize the need for vigilant surveillance at mass gatherings.IMPORTANCE Large population sizes and densities at mass gatherings such as the Hajj (Makkah, Saudi Arabia) can contribute to outbreaks of respiratory virus infection by providing local hot spots for transmission followed by spread to other localities. Using a genome-scale analysis, we show that the genetic diversity of influenza A viruses at the Hajj gatherings during 2013 to 2015 was largely shaped by the introduction of multiple viruses from diverse geographic regions, including the Middle East, with only little evidence of interhost virus transmission at the Hajj and seemingly limited spread of subconsensus mutational variants. The diversity of viruses at the Hajj pilgrimages highlights the potential for lineage cocirculation during mass gatherings, in turn fuelling segment reassortment and the emergence of novel variants, such that the continued surveillance of respiratory pathogens at mass gatherings should be a public health priority.
Keywords: Hajj; epidemiology; evolution; influenza; phylogeny; transmission.
Copyright © 2017 American Society for Microbiology.
Figures

Similar articles
-
Mass Gatherings and the Spread of Respiratory Infections. Lessons from the Hajj.Ann Am Thorac Soc. 2016 Jun;13(6):759-65. doi: 10.1513/AnnalsATS.201511-772FR. Ann Am Thorac Soc. 2016. PMID: 27088298
-
Influenza not MERS CoV among returning Hajj and Umrah pilgrims with respiratory illness, Kashmir, north India, 2014-15.Travel Med Infect Dis. 2017 Jan-Feb;15:45-47. doi: 10.1016/j.tmaid.2016.12.002. Epub 2016 Dec 6. Travel Med Infect Dis. 2017. PMID: 27932291 Free PMC article.
-
Active screening and surveillance in the United Kingdom for Middle East respiratory syndrome coronavirus in returning travellers and pilgrims from the Middle East: a prospective descriptive study for the period 2013-2015.Int J Infect Dis. 2016 Jun;47:10-4. doi: 10.1016/j.ijid.2016.04.016. Epub 2016 Apr 23. Int J Infect Dis. 2016. PMID: 27117200 Free PMC article.
-
Respiratory tract infections during the annual Hajj: potential risks and mitigation strategies.Curr Opin Pulm Med. 2013 May;19(3):192-7. doi: 10.1097/MCP.0b013e32835f1ae8. Curr Opin Pulm Med. 2013. PMID: 23429098 Review.
-
Establishment of public health security in Saudi Arabia for the 2009 Hajj in response to pandemic influenza A H1N1.Lancet. 2009 Nov 21;374(9703):1786-91. doi: 10.1016/S0140-6736(09)61927-9. Epub 2009 Nov 14. Lancet. 2009. PMID: 19914707 Review.
Cited by
-
Genome characterization and mutation analysis of human influenza A virus in Thailand.Genomics Inform. 2022 Jun;20(2):e21. doi: 10.5808/gi.21077. Epub 2022 Jun 30. Genomics Inform. 2022. PMID: 35794701 Free PMC article.
-
Genetic Diversity and Evolutionary Kinetics of Influenza A Virus H3N2 Subtypes Circulating in Riyadh, Saudi Arabia.Vaccines (Basel). 2023 Mar 20;11(3):702. doi: 10.3390/vaccines11030702. Vaccines (Basel). 2023. PMID: 36992286 Free PMC article.
-
Patients with influenza admitted to a tertiary-care hospital in Riyadh between 2018 and 2022: characteristics, outcomes and factors associated with ICU admission and mortality.BMC Pulm Med. 2024 Sep 19;24(1):464. doi: 10.1186/s12890-024-03281-6. BMC Pulm Med. 2024. PMID: 39300448 Free PMC article.
-
Epidemiological Differences in Hajj-Acquired Airborne Infections in Pilgrims Arriving from Low and Middle-Income versus High-Income Countries: A Systematised Review.Trop Med Infect Dis. 2023 Aug 17;8(8):418. doi: 10.3390/tropicalmed8080418. Trop Med Infect Dis. 2023. PMID: 37624356 Free PMC article. Review.
-
Healthcare Research in Mass Religious Gatherings and Emergency Management: A Comprehensive Narrative Review.Healthcare (Basel). 2023 Jan 13;11(2):244. doi: 10.3390/healthcare11020244. Healthcare (Basel). 2023. PMID: 36673612 Free PMC article. Review.
References
-
- Al-Tawfiq JA, Memish ZA. 2012. The Hajj: updated health hazards and current recommendations for 2012. Euro Surveill 17(41):pii=20295 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20295. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
