

Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia
- PMID: 28331220
- PMCID: PMC5537416
- DOI: 10.1038/jhg.2017.38
Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia
Abstract
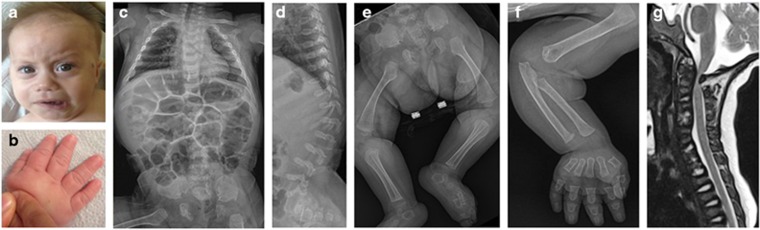
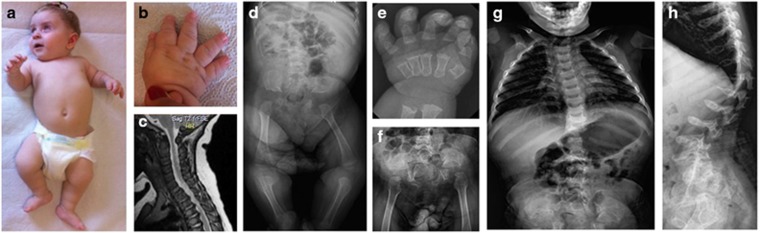
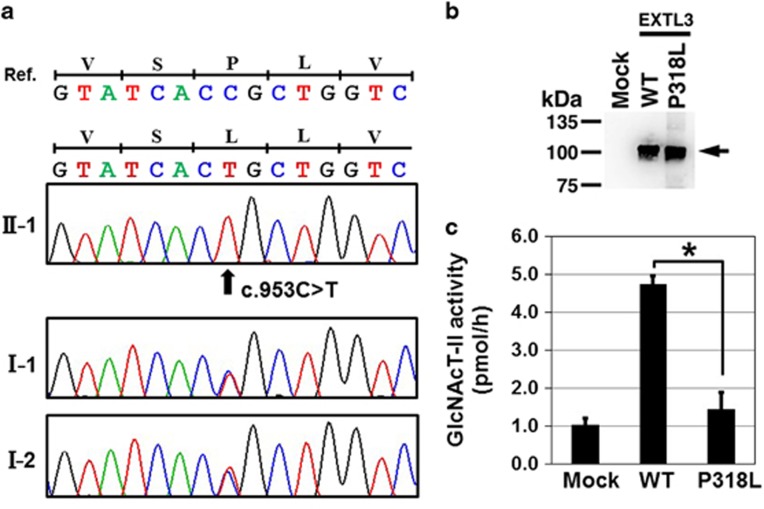
Spondylo-epi-metaphyseal dysplasia (SEMD) is a group of inherited skeletal diseases characterized by the anomalies in spine, epiphyses and metaphyses. SEMD is highly heterogeneous and >20 distinct entities have been identified. Here we describe a novel type of SEMD in two unrelated Turkish patients who presented with severe platyspondyly, kyphoscoliosis, pelvic distortion, constriction of the proximal femora and brachydactyly. Although these phenotypes overlap considerably with some known SEMDs, they had a novel causal gene, exostosin-like glycosyltransferase 3 (EXTL3), that encodes a glycosyltransferase involved in the synthesis of heparin and heparan sulfate. The EXTL3 mutation identified in the patients was a homozygous missense mutation (c.953C>T) that caused a substitution in a highly conserved amino acid (p.P318L). The enzyme activity of the mutant EXTL3 protein was significantly decreased compared to the wild-type protein. Both patients had spinal cord compression at the cranio-vertebral junction and multiple liver cysts since early infancy. One of the patients showed severe immunodeficiency, which is considered non-fortuitous association. Our findings would help define a novel type of SEMD caused by EXTL3 mutations.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Guo, L., Elcioglu, N. H., Iida, A., Demirkol, Y. K., Aras, S., Matsumoto, N. et al. Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J. Hum. Genet. 62, 447–451 (2016). - PubMed
-
- Guo, L., Girisha, K. M., Iida, A., Hebbar, M., Shukla, A., Shah, H. et al. Identification of a novel LRRK1 mutation in a family with osteosclerotic metaphyseal dysplasia. J. Hum. Genet. 62, 437–441 (2016). - PubMed
-
- Wang, Z., Horemuzova, E., Iida, A., Guo, L., Liu, Y., Matsumoto, N. et al. Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J. Hum. Genet. (e-pub ahead of print 26 January 2017; doi: 10.1038/jhg.2016.157). - PubMed
-
- Miyatake, S., Tada, H., Moriya, S., Takanashi, J., Hirano, Y., Hayashi, M. et al. Atypical giant axonal neuropathy arising from a homozygous mutation by uniparental isodisomy. Clin. Genet. 87, 395–397 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials