

Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles
- PMID: 28333380
- PMCID: PMC5575875
- DOI: 10.1002/cphy.c160011
Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles
Abstract
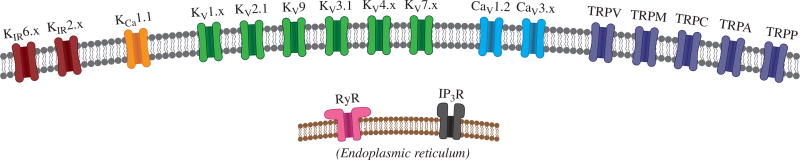
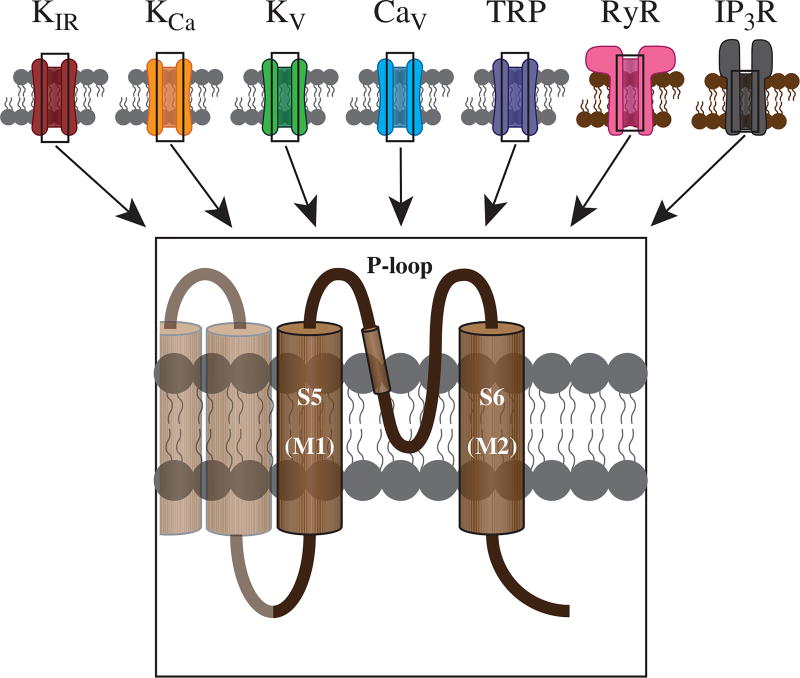
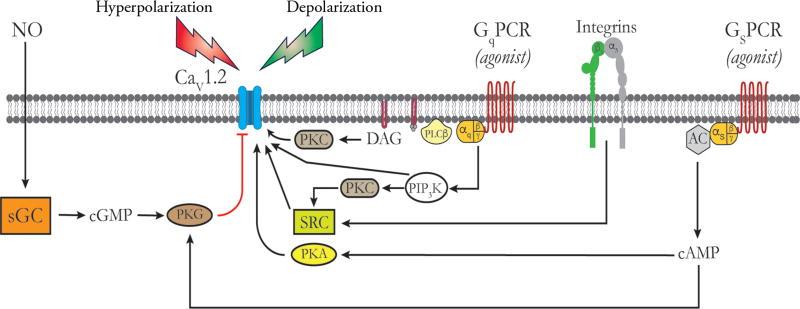
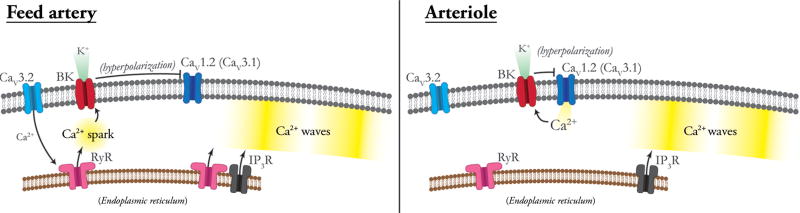
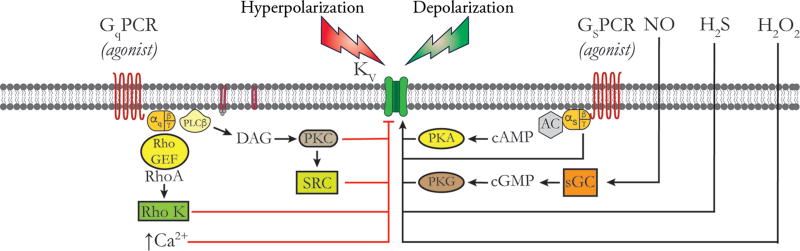
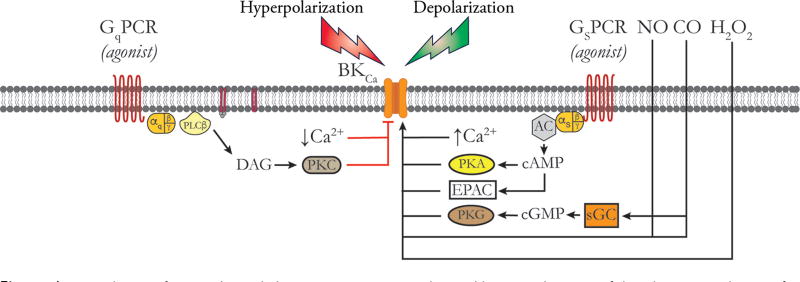
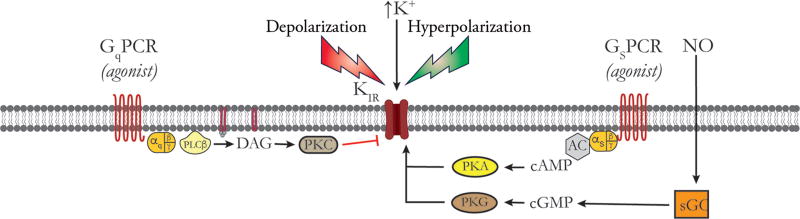
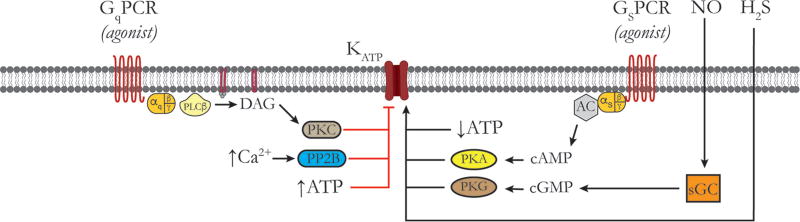
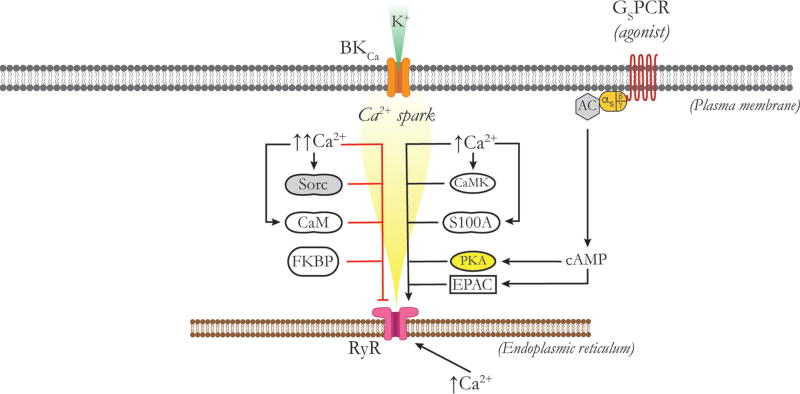
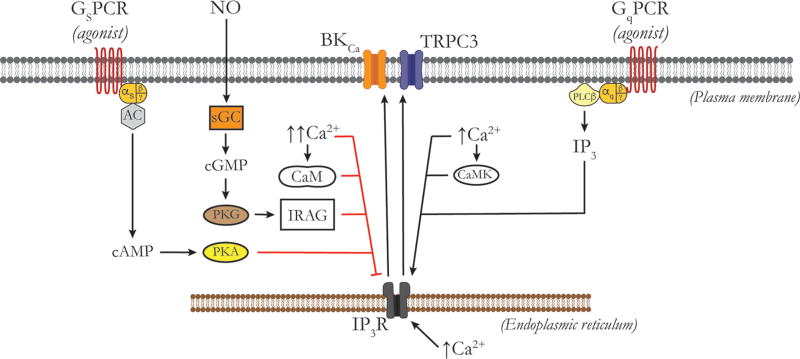
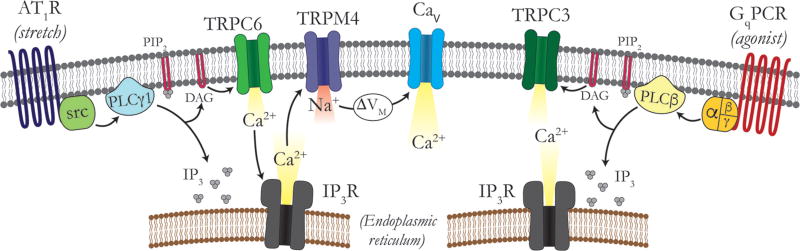
Vascular tone of resistance arteries and arterioles determines peripheral vascular resistance, contributing to the regulation of blood pressure and blood flow to, and within the body's tissues and organs. Ion channels in the plasma membrane and endoplasmic reticulum of vascular smooth muscle cells (SMCs) in these blood vessels importantly contribute to the regulation of intracellular Ca2+ concentration, the primary determinant of SMC contractile activity and vascular tone. Ion channels provide the main source of activator Ca2+ that determines vascular tone, and strongly contribute to setting and regulating membrane potential, which, in turn, regulates the open-state-probability of voltage gated Ca2+ channels (VGCCs), the primary source of Ca2+ in resistance artery and arteriolar SMCs. Ion channel function is also modulated by vasoconstrictors and vasodilators, contributing to all aspects of the regulation of vascular tone. This review will focus on the physiology of VGCCs, voltage-gated K+ (KV) channels, large-conductance Ca2+-activated K+ (BKCa) channels, strong-inward-rectifier K+ (KIR) channels, ATP-sensitive K+ (KATP) channels, ryanodine receptors (RyRs), inositol 1,4,5-trisphosphate receptors (IP3Rs), and a variety of transient receptor potential (TRP) channels that contribute to pressure-induced myogenic tone in resistance arteries and arterioles, the modulation of the function of these ion channels by vasoconstrictors and vasodilators, their role in the functional regulation of tissue blood flow and their dysfunction in diseases such as hypertension, obesity, and diabetes. © 2017 American Physiological Society. Compr Physiol 7:485-581, 2017.
Copyright © 2017 John Wiley & Sons, Inc.
Figures
References
-
- Abbink EJ, Wollersheim H, Netten PM, Russel FG, Lutterman JA, Smits P. Microcirculatory effects of KATP channel blockade by sulphonylurea derivatives in humans. Eur J Clin Invest. 2002;32:163–171. - PubMed
-
- Abdel-Latif AA. Cross talk between cyclic nucleotides and polyphos-phoinositide hydrolysis, protein kinases, and contraction in smooth muscle. Exp Biol Med (Maywood) 2001;226:153–163. - PubMed
-
- Abdi A, Mazzocco C, Legeron FP, Yvert B, Macrez N, Morel JL. TRPP2 modulates ryanodine- and inositol-1,4,5-trisphosphate receptors-dependent Ca2+ signals in opposite ways in cerebral arteries. Cell Calcium. 2015;58:467–475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
