

Pseudoalignment for metagenomic read assignment
- PMID: 28334086
- PMCID: PMC5870846
- DOI: 10.1093/bioinformatics/btx106
Pseudoalignment for metagenomic read assignment
Abstract
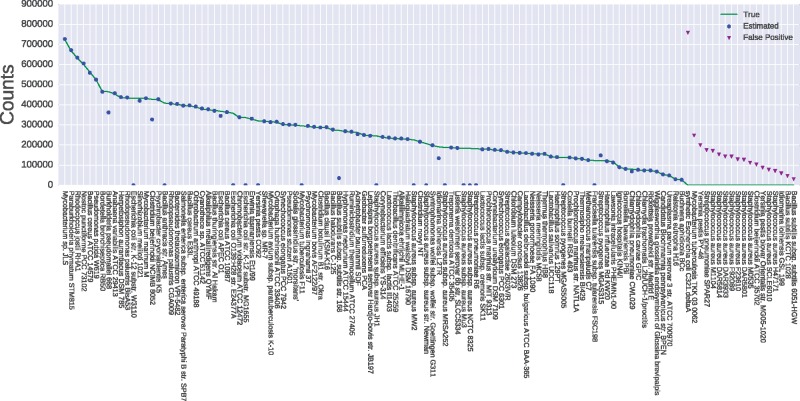
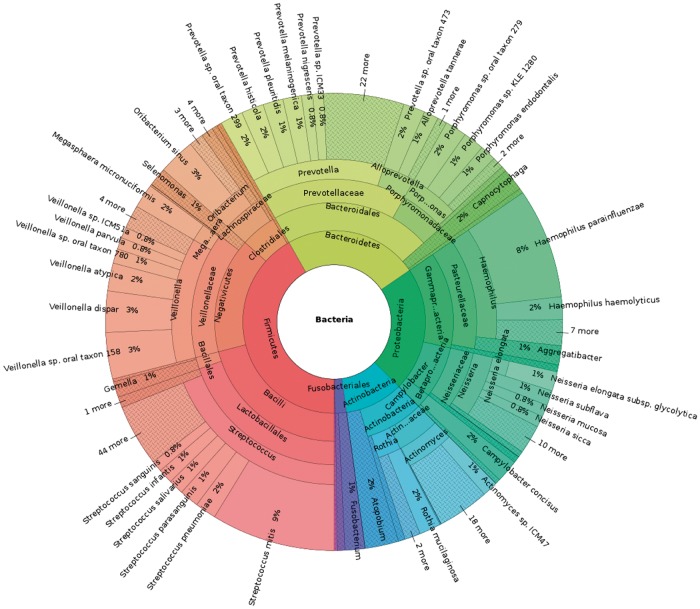
Motivation: Read assignment is an important first step in many metagenomic analysis workflows, providing the basis for identification and quantification of species. However ambiguity among the sequences of many strains makes it difficult to assign reads at the lowest level of taxonomy, and reads are typically assigned to taxonomic levels where they are unambiguous. We explore connections between metagenomic read assignment and the quantification of transcripts from RNA-Seq data in order to develop novel methods for rapid and accurate quantification of metagenomic strains.
Results: We find that the recent idea of pseudoalignment introduced in the RNA-Seq context is highly applicable in the metagenomics setting. When coupled with the Expectation-Maximization (EM) algorithm, reads can be assigned far more accurately and quickly than is currently possible with state of the art software, making it possible and practical for the first time to analyze abundances of individual genomes in metagenomics projects.
Availability and implementation: Pipeline and analysis code can be downloaded from http://github.com/pachterlab/metakallisto.
Contact: lpachter@math.berkeley.edu.
© The Author (2017). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com
Figures

References
-
- Bray N. et al. (2015). Near-optimal RNA-Seq quantification. Nature Biotechnol, 34, 525–527. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
