

Strategies for processing and quality control of Illumina genotyping arrays
- PMID: 28334151
- PMCID: PMC6171493
- DOI: 10.1093/bib/bbx012
Strategies for processing and quality control of Illumina genotyping arrays
Abstract
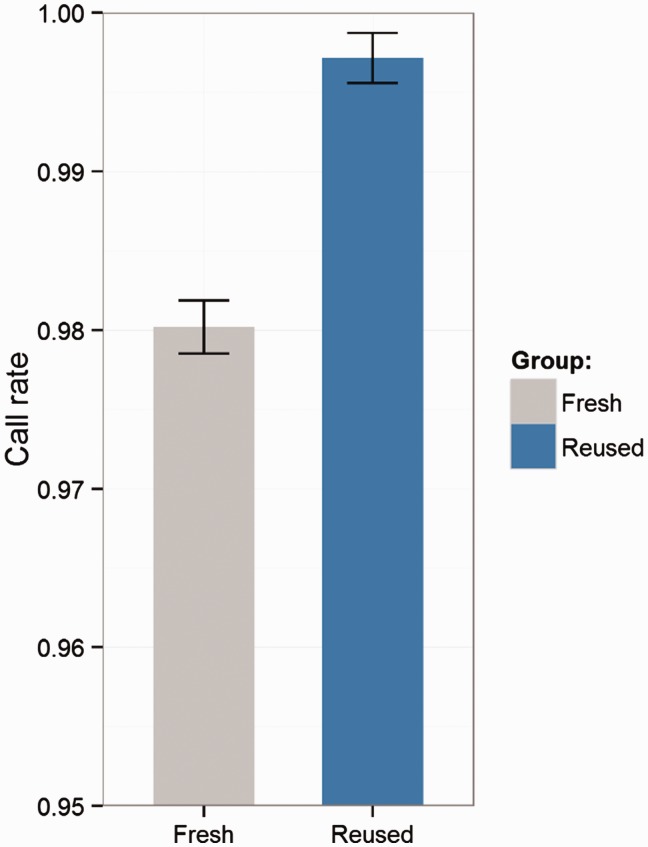
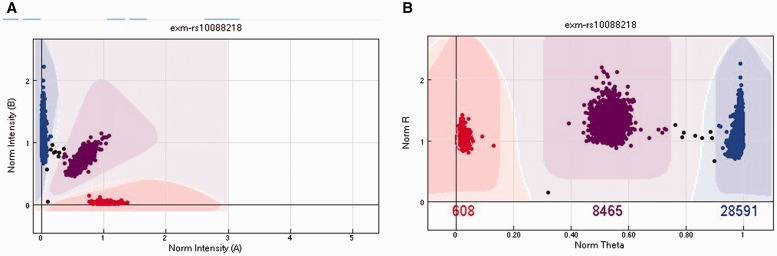
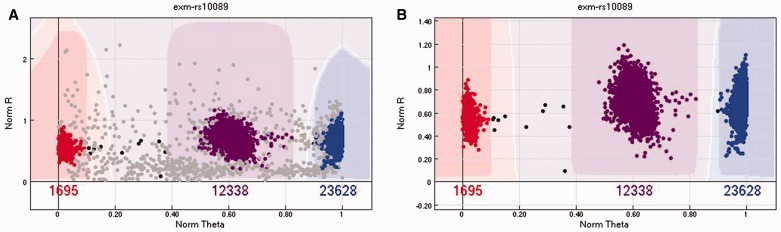
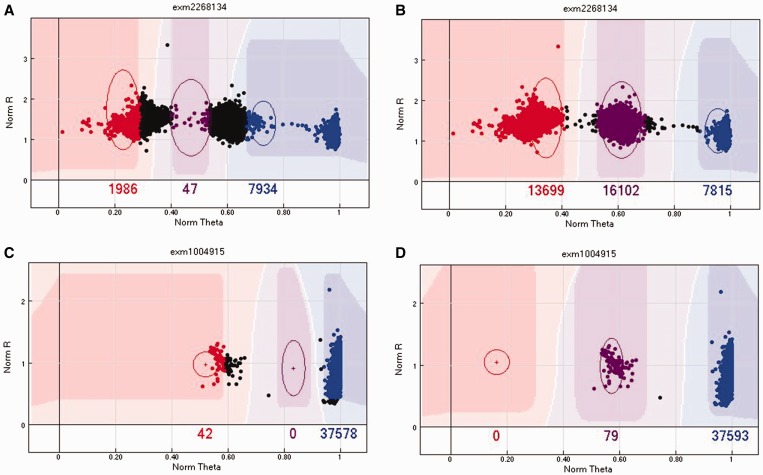
Illumina genotyping arrays have powered thousands of large-scale genome-wide association studies over the past decade. Yet, because of the tremendous volume and complicated genetic assumptions of Illumina genotyping data, processing and quality control (QC) of these data remain a challenge. Thorough QC ensures the accurate identification of single-nucleotide polymorphisms and is required for the correct interpretation of genetic association results. By processing genotyping data on > 100 000 subjects from >10 major Illumina genotyping arrays, we have accumulated extensive experience in handling some of the most peculiar scenarios related to the processing and QC of Illumina genotyping data. Here, we describe strategies for processing Illumina genotyping data from the raw data to an analysis ready format, and we elaborate on the necessary QC procedures required at each processing step. High-quality Illumina genotyping data sets can be obtained by following our detailed QC strategies.
Figures
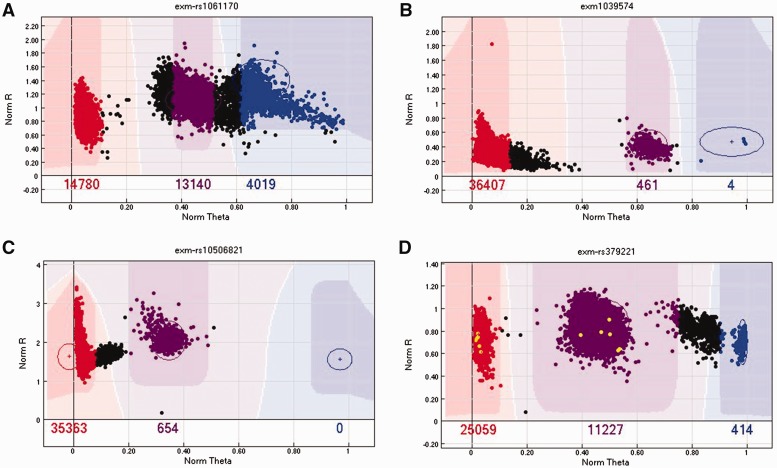
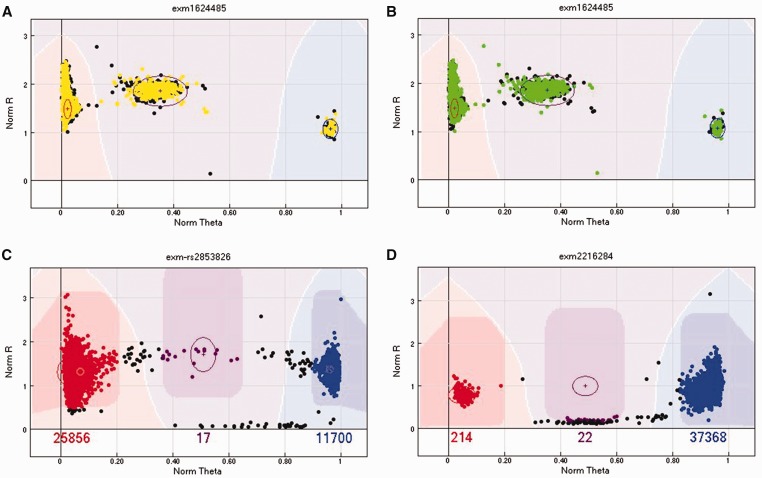
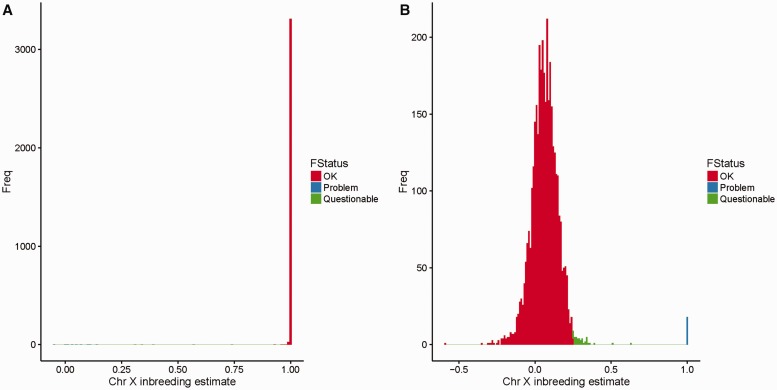
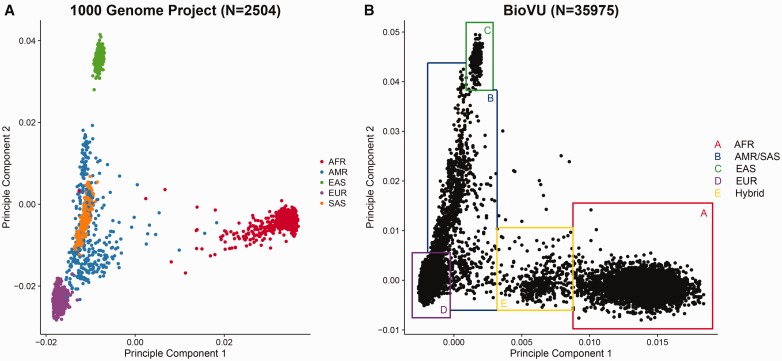
References
-
- Cloonan N, Forrest AR, Kolle G, et al.Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 2008;5:613–19. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
