

In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases
- PMID: 28334821
- PMCID: PMC5416837
- DOI: 10.1093/nar/gkx116
In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases
Abstract
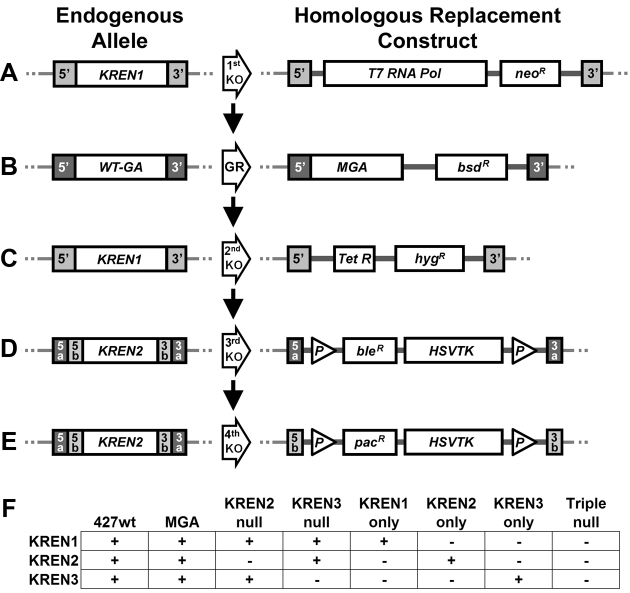
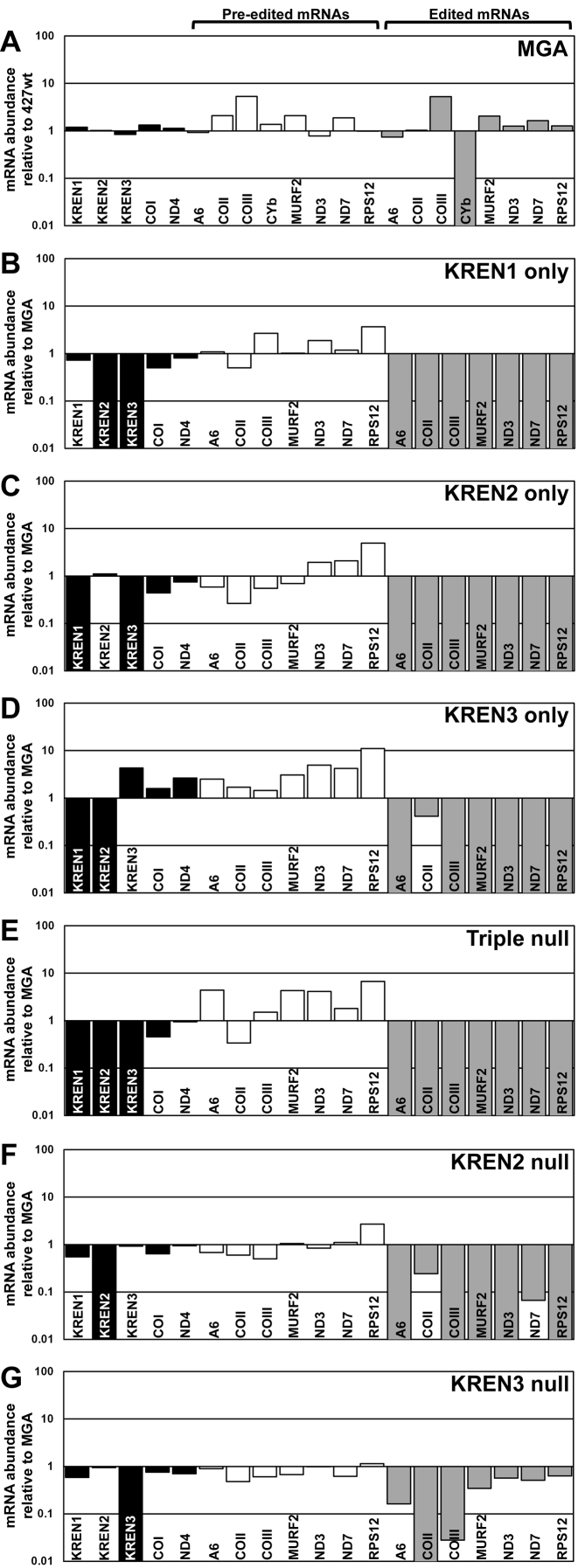
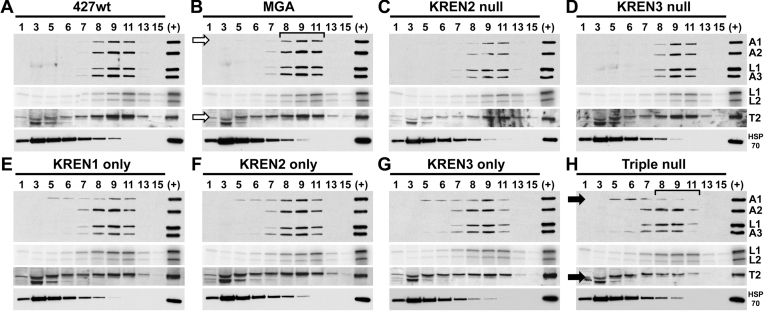
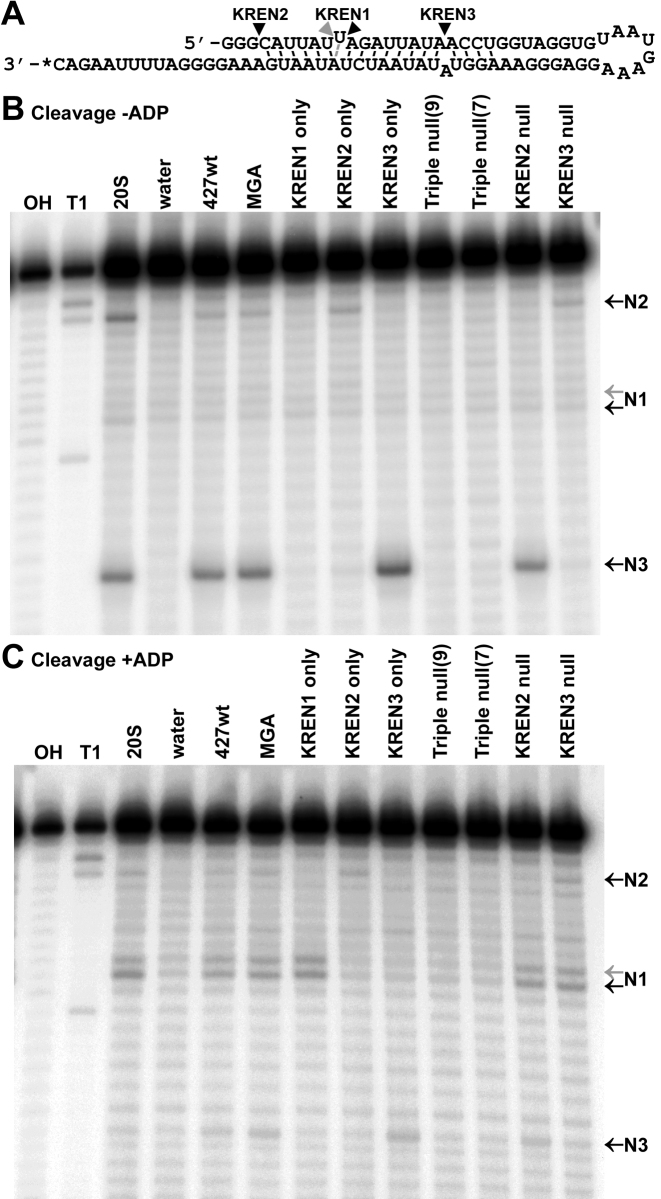
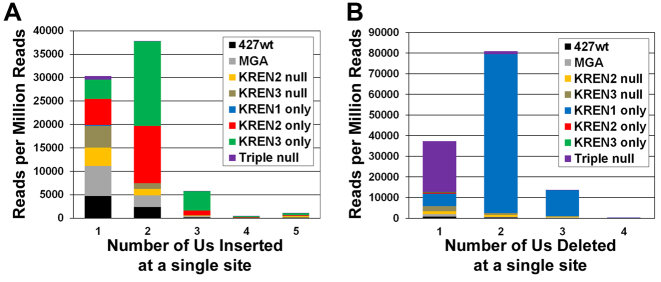
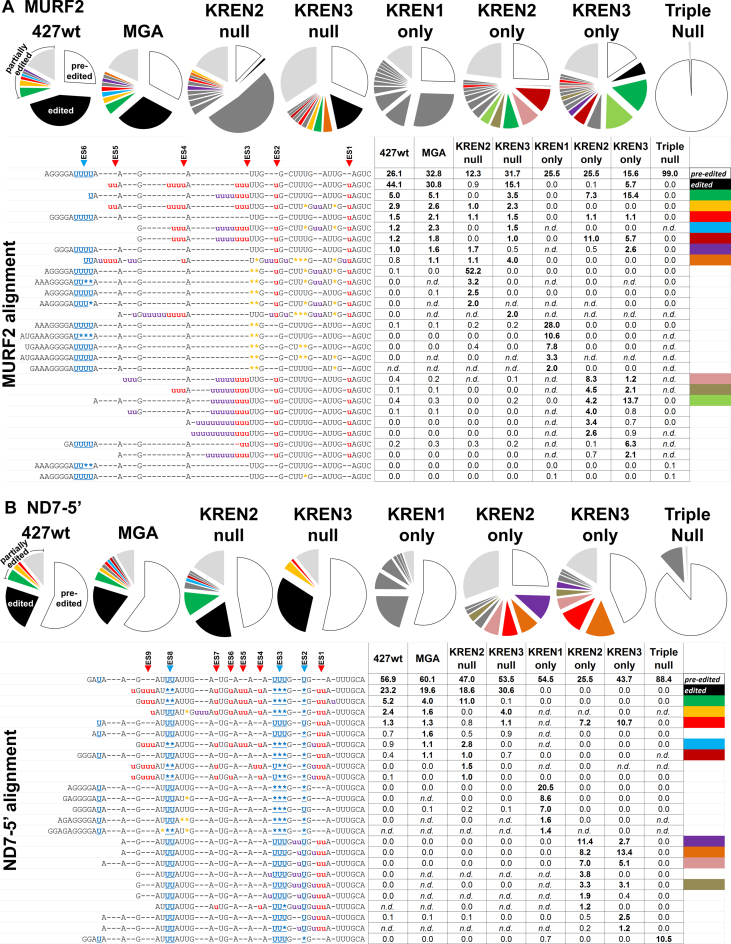
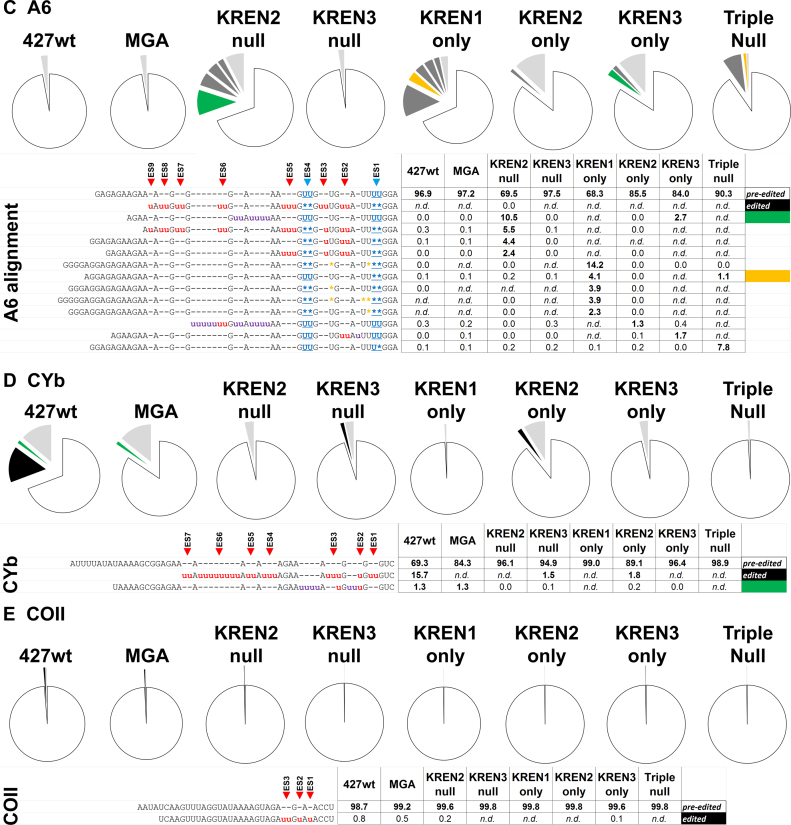
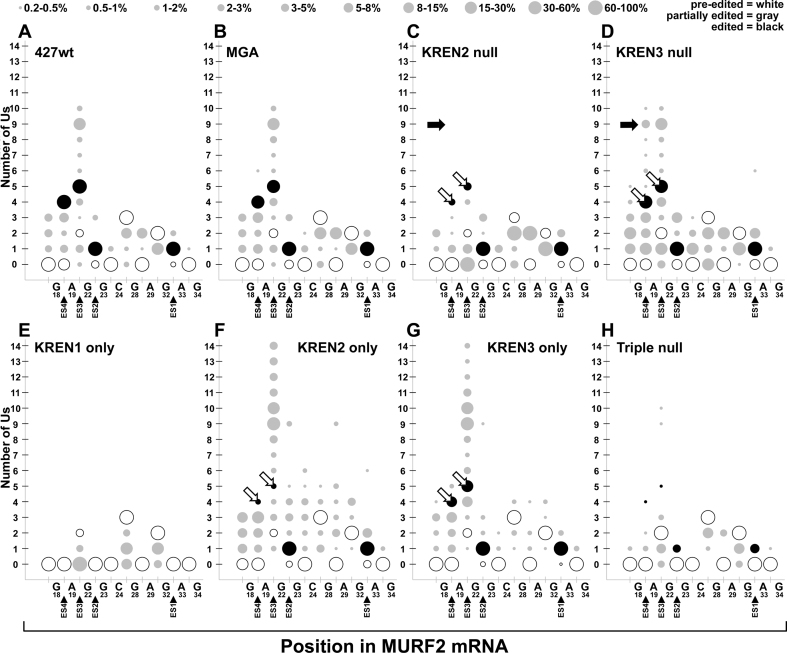
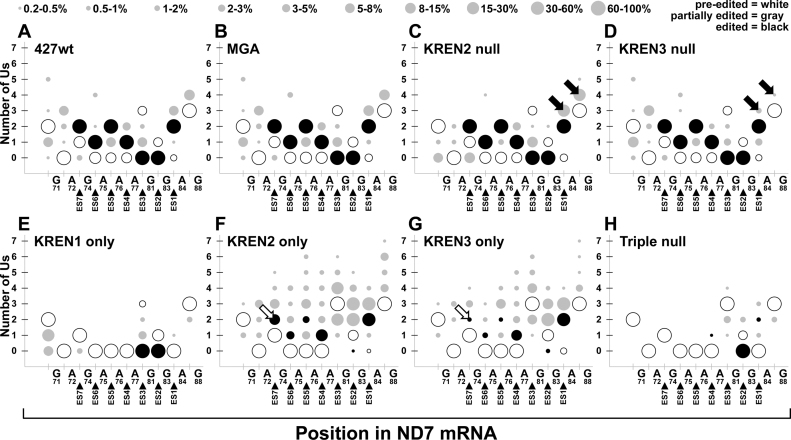
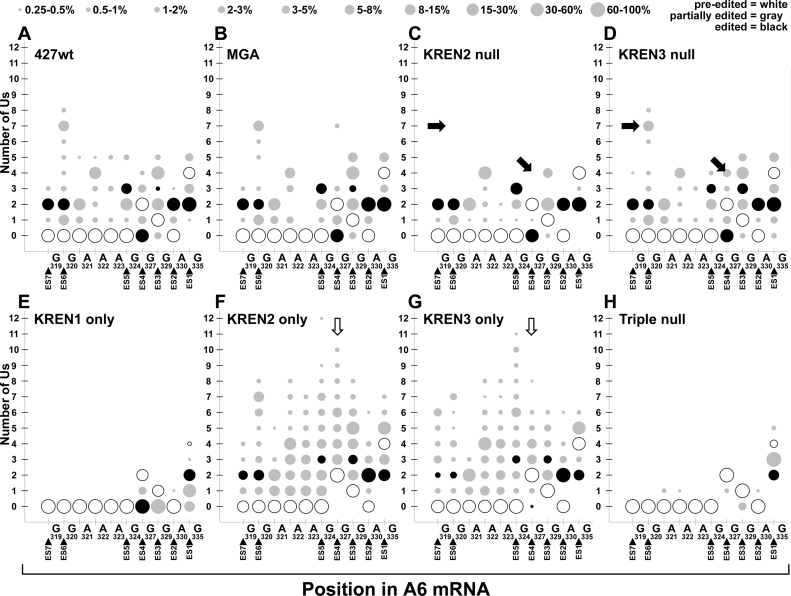
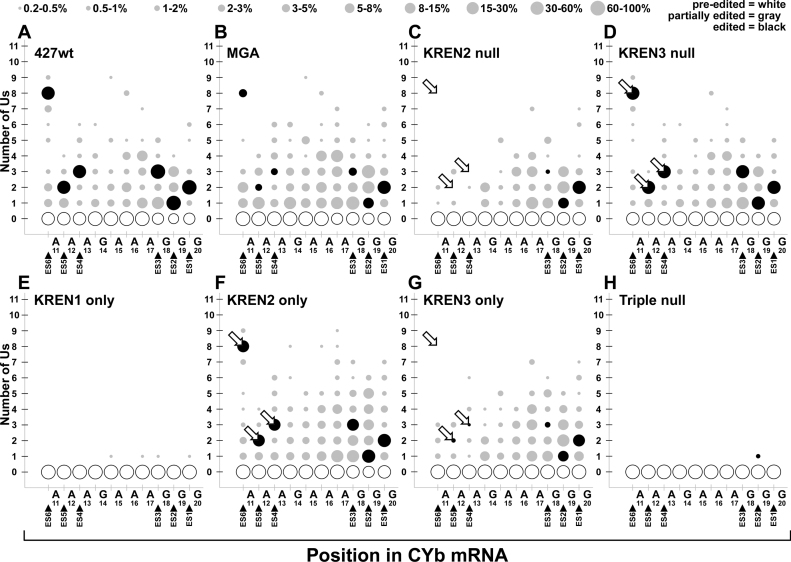
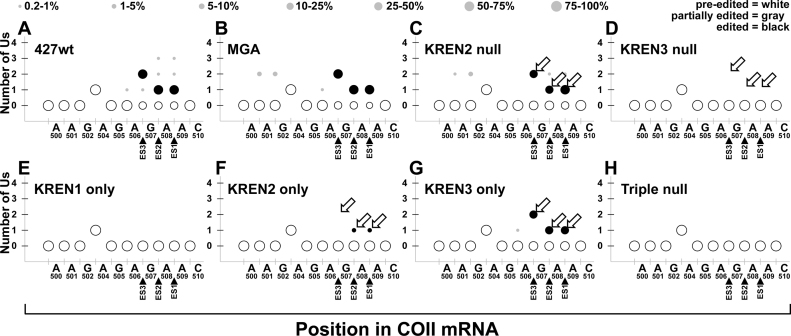
RNA editing is an essential post-transcriptional process that creates functional mitochondrial mRNAs in Kinetoplastids. Multiprotein editosomes catalyze pre-mRNA cleavage, uridine (U) insertion or deletion, and ligation as specified by guide RNAs. Three functionally and compositionally distinct editosomes differ by the mutually exclusive presence of the KREN1, KREN2 or KREN3 endonuclease and their associated partner proteins. Because endonuclease cleavage is a likely point of regulation for RNA editing, we elucidated endonuclease specificity in vivo. We used a mutant gamma ATP synthase allele (MGA) to circumvent the normal essentiality of the editing endonucleases, and created cell lines in which both alleles of one, two or all three of the endonucleases were deleted. Cells lacking multiple endonucleases had altered editosome sedimentation on glycerol gradients and substantial defects in overall editing. Deep sequencing analysis of RNAs from such cells revealed clear discrimination by editosomes between sites of deletion versus insertion editing and preferential but overlapping specificity for sites of insertion editing. Thus, endonuclease specificities in vivo are distinct but with some functional overlap. The overlapping specificities likely accommodate the more numerous sites of insertion versus deletion editing as editosomes collaborate to accurately edit thousands of distinct editing sites in vivo.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Editosome RNase III domain interactions are essential for editing and differ between life cycle stages in Trypanosoma brucei.RNA. 2019 Sep;25(9):1150-1163. doi: 10.1261/rna.071258.119. Epub 2019 Jun 6. RNA. 2019. PMID: 31171708 Free PMC article.
-
Endonuclease associations with three distinct editosomes in Trypanosoma brucei.J Biol Chem. 2011 Jun 3;286(22):19320-30. doi: 10.1074/jbc.M111.228965. Epub 2011 Apr 7. J Biol Chem. 2011. PMID: 21474442 Free PMC article.
-
KREPB6, KREPB7, and KREPB8 are important for editing endonuclease function in Trypanosoma brucei.RNA. 2012 Feb;18(2):308-20. doi: 10.1261/rna.029314.111. Epub 2011 Dec 19. RNA. 2012. PMID: 22184461 Free PMC article.
-
Mitochondrial RNA editing in trypanosomes: small RNAs in control.Biochimie. 2014 May;100:125-31. doi: 10.1016/j.biochi.2014.01.003. Epub 2014 Jan 17. Biochimie. 2014. PMID: 24440637 Free PMC article. Review.
-
Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer.Wiley Interdiscip Rev RNA. 2011 Sep-Oct;2(5):669-85. doi: 10.1002/wrna.82. Epub 2011 Mar 23. Wiley Interdiscip Rev RNA. 2011. PMID: 21823228 Free PMC article. Review.
Cited by
-
Trypanosome RNAEditing Substrate Binding Complex integrity and function depends on the upstream action of RESC10.Nucleic Acids Res. 2021 Apr 6;49(6):3557-3572. doi: 10.1093/nar/gkab129. Nucleic Acids Res. 2021. PMID: 33677542 Free PMC article.
-
Domain function and predicted structure of three heterodimeric endonuclease subunits of RNA editing catalytic complexes in Trypanosoma brucei.Nucleic Acids Res. 2022 Sep 23;50(17):10123-10139. doi: 10.1093/nar/gkac753. Nucleic Acids Res. 2022. PMID: 36095119 Free PMC article.
-
KREH1 RNA helicase activity promotes utilization of initiator gRNAs across multiple mRNAs in trypanosome RNA editing.Nucleic Acids Res. 2023 Jun 23;51(11):5791-5809. doi: 10.1093/nar/gkad292. Nucleic Acids Res. 2023. PMID: 37140035 Free PMC article.
-
Site-specific and substrate-specific control of accurate mRNA editing by a helicase complex in trypanosomes.RNA. 2020 Dec;26(12):1862-1881. doi: 10.1261/rna.076513.120. Epub 2020 Sep 1. RNA. 2020. PMID: 32873716 Free PMC article.
-
Complete minicircle genome of Leptomonas pyrrhocoris reveals sources of its non-canonical mitochondrial RNA editing events.Nucleic Acids Res. 2021 Apr 6;49(6):3354-3370. doi: 10.1093/nar/gkab114. Nucleic Acids Res. 2021. PMID: 33660779 Free PMC article.
References
-
- Stuart K.D., Schnaufer A., Ernst N.L., Panigrahi A.K.. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 2005; 30:97–105. - PubMed
-
- Schnaufer A., Domingo G.J., Stuart K.D.. Natural and induced dyskinetoplastid trypanosomatids: how to live without mitochondrial DNA. Int. J. Parasitol. 2002; 32:1071–1084. - PubMed
-
- Trotter J.R., Ernst N.L., Carnes J., Panicucci B., Stuart K.. A deletion site editing endonuclease in Trypanosoma brucei. Mol. Cell. 2005; 20:403–412. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
